

A role for circular code properties in translation
- PMID: 33911089
- PMCID: PMC8080828
- DOI: 10.1038/s41598-021-87534-y
A role for circular code properties in translation
Abstract
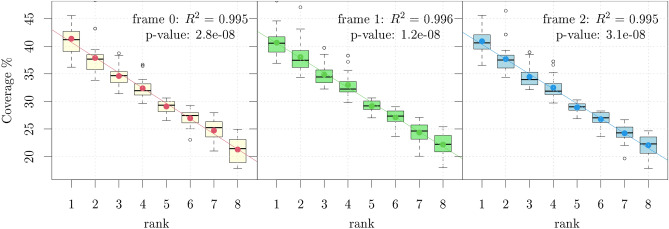
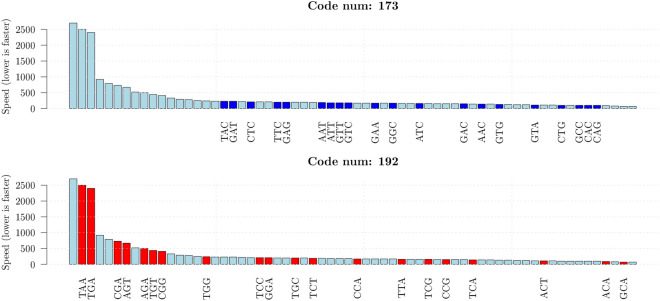
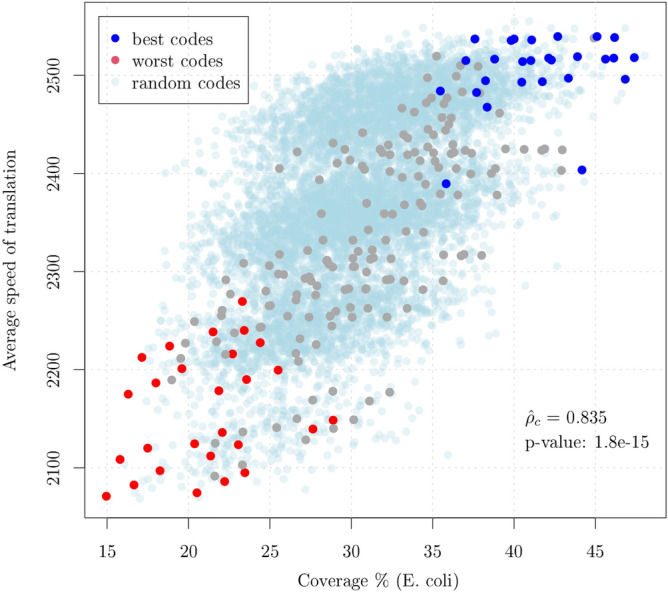
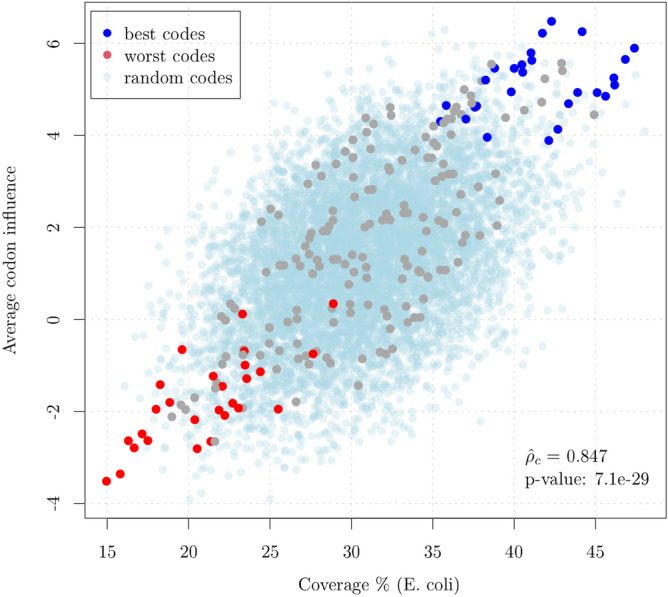
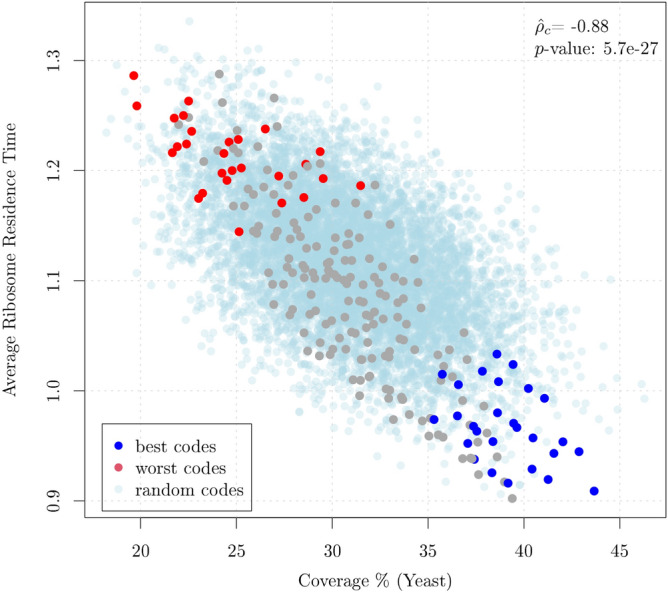
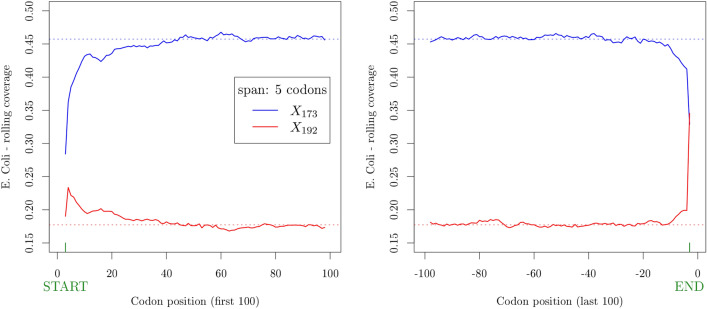
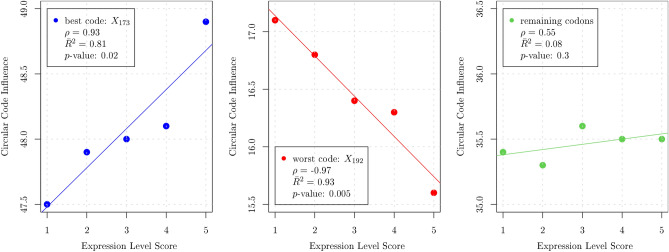
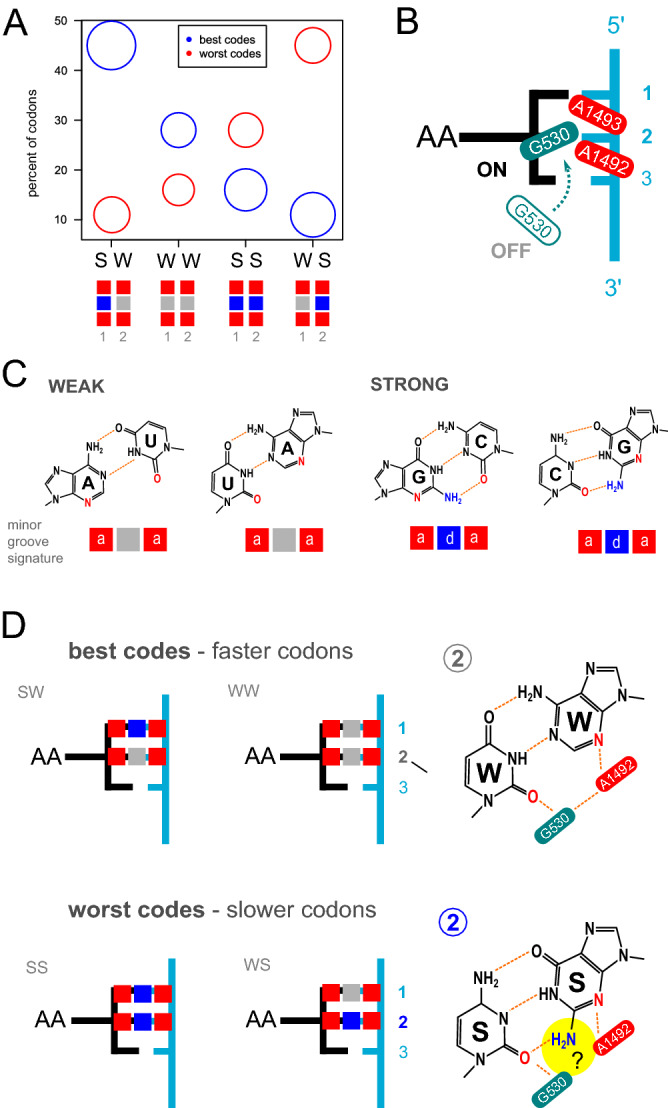
Circular codes represent a form of coding allowing detection/correction of frame-shift errors. Building on recent theoretical advances on circular codes, we provide evidence that protein coding sequences exhibit in-frame circular code marks, that are absent in introns and are intimately linked to the keto-amino transformation of codon bases. These properties strongly correlate with translation speed, codon influence and protein synthesis levels. Strikingly, circular code marks are absent at the beginning of coding sequences, but stably occur 40 codons after the initiator codon, hinting at the translation elongation process. Finally, we use the lens of circular codes to show that codon influence on translation correlates with the strong-weak dichotomy of the first two bases of the codon. The results can lead to defining new universal tools for sequence indicators and sequence optimization for bioinformatics and biotechnological applications, and can shed light on the molecular mechanisms behind the decoding process.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
