

Towards complete and error-free genome assemblies of all vertebrate species
- PMID: 33911273
- PMCID: PMC8081667
- DOI: 10.1038/s41586-021-03451-0
Towards complete and error-free genome assemblies of all vertebrate species
Abstract
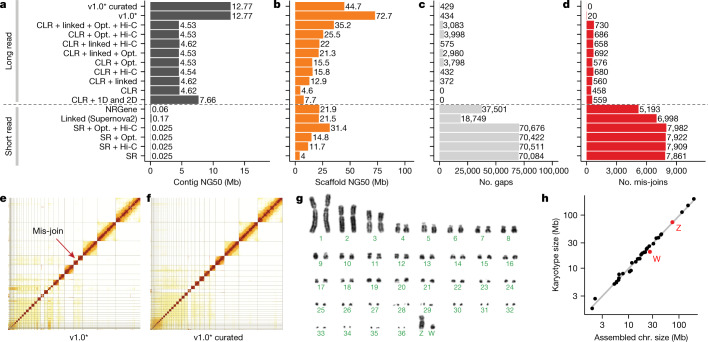
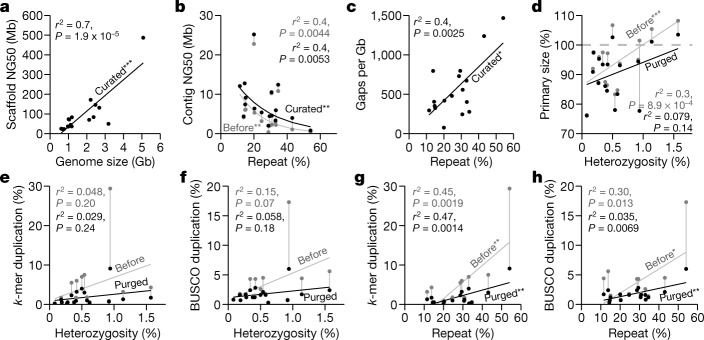
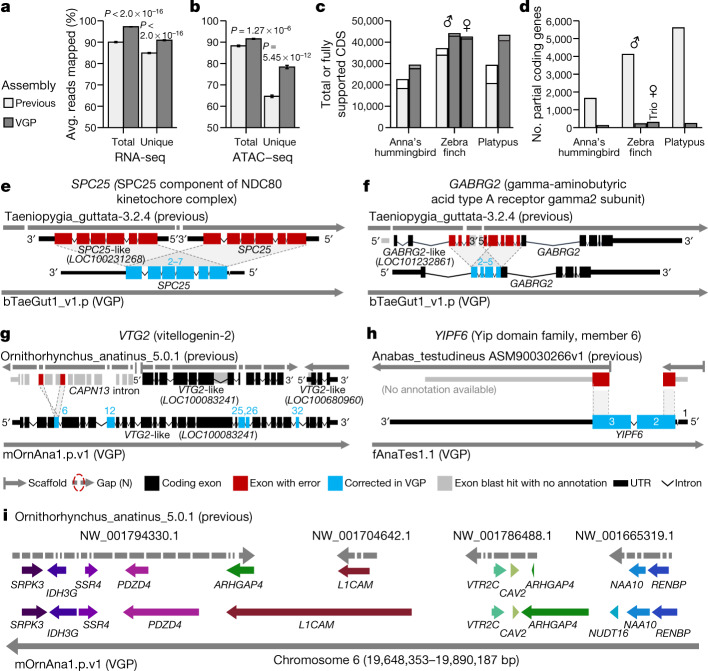
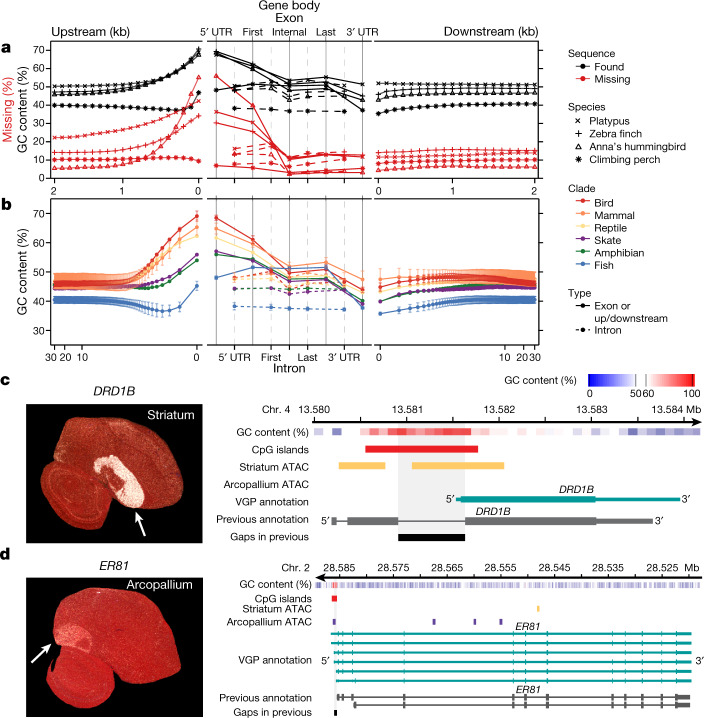
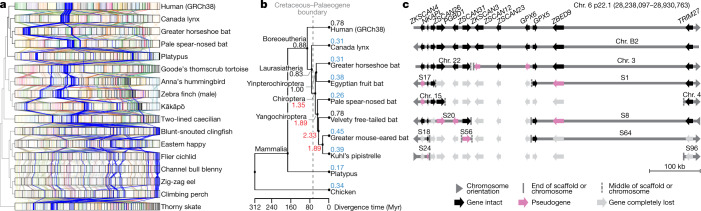
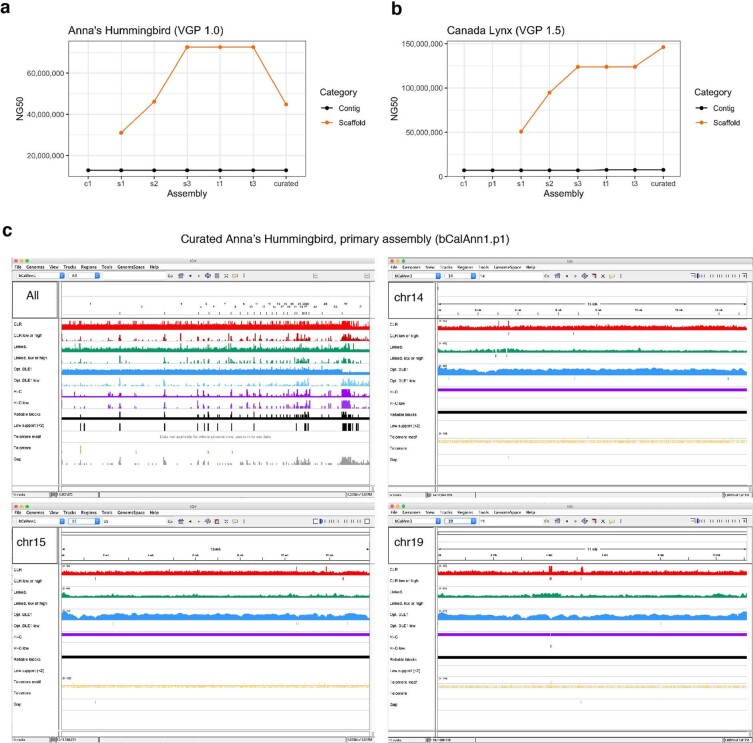
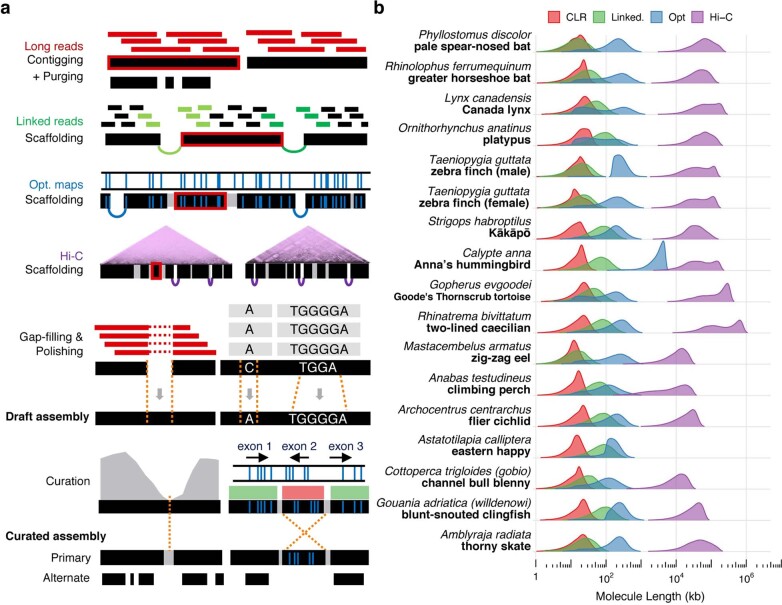
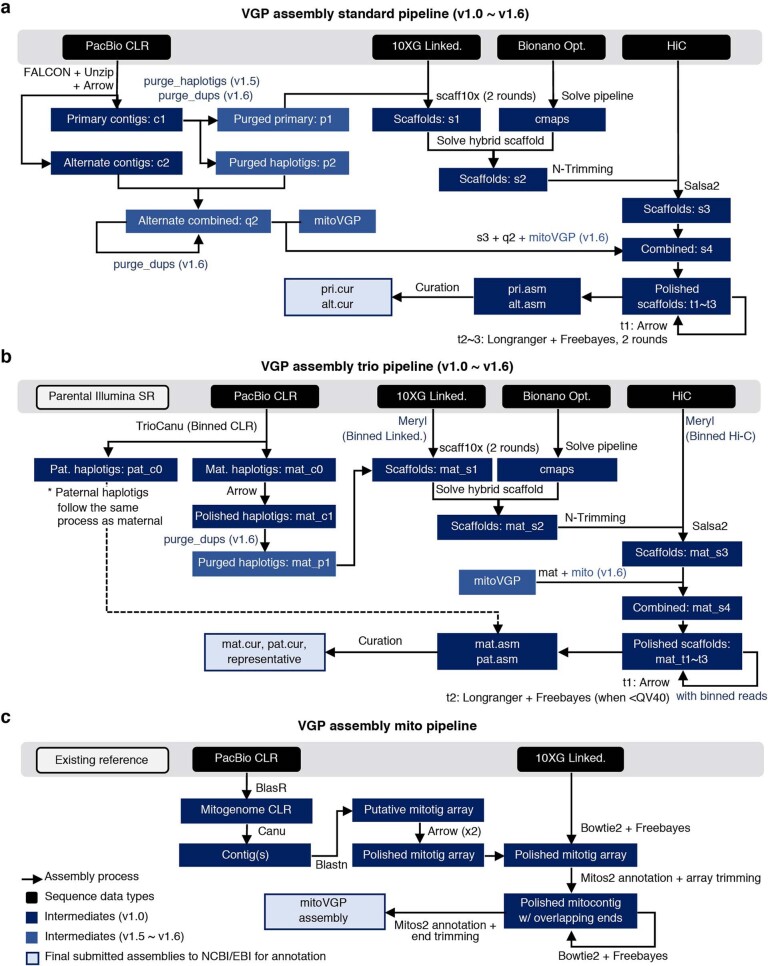
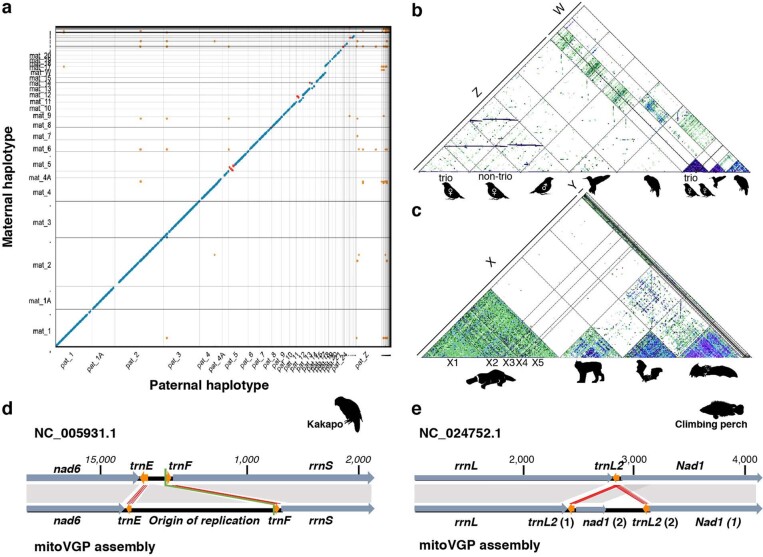
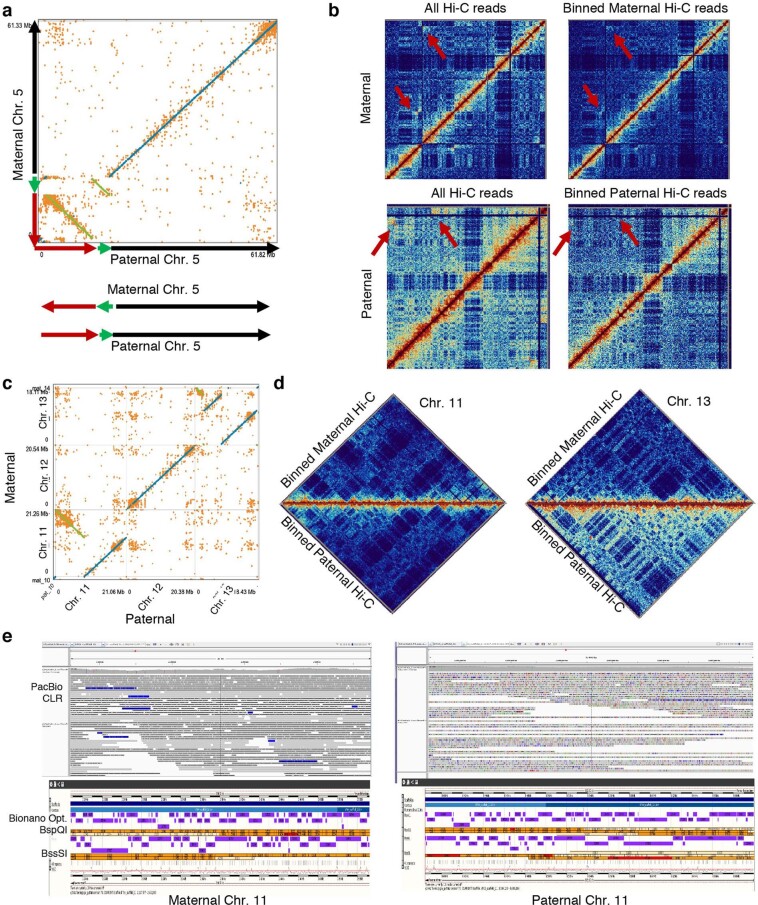
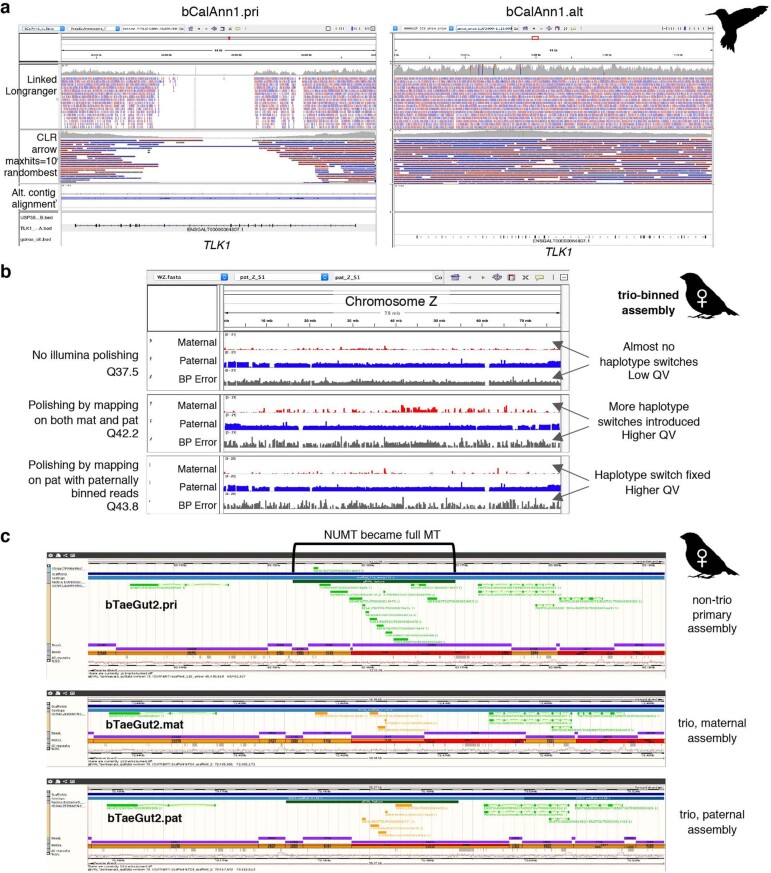
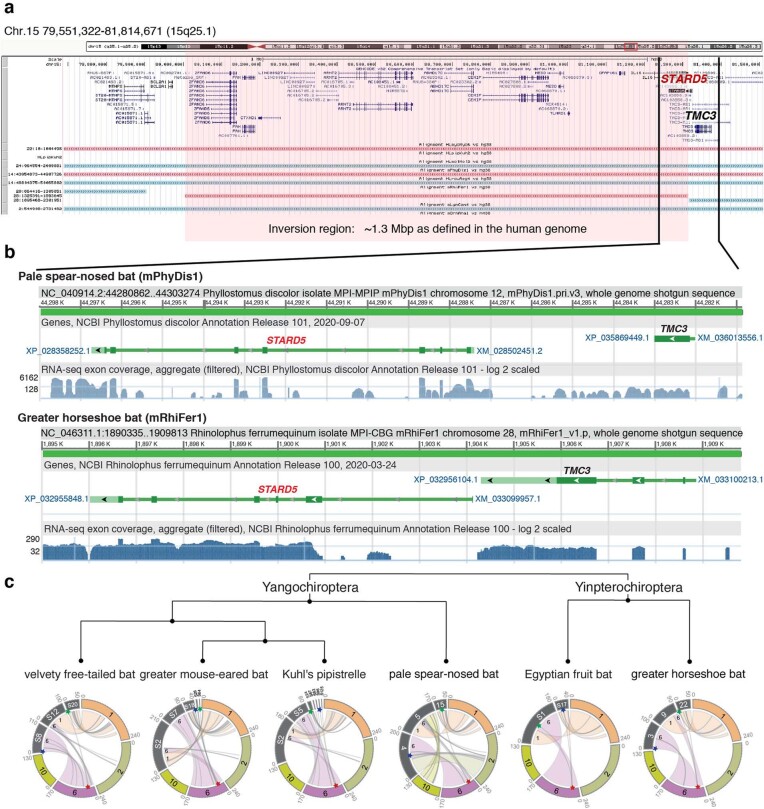
High-quality and complete reference genome assemblies are fundamental for the application of genomics to biology, disease, and biodiversity conservation. However, such assemblies are available for only a few non-microbial species1-4. To address this issue, the international Genome 10K (G10K) consortium5,6 has worked over a five-year period to evaluate and develop cost-effective methods for assembling highly accurate and nearly complete reference genomes. Here we present lessons learned from generating assemblies for 16 species that represent six major vertebrate lineages. We confirm that long-read sequencing technologies are essential for maximizing genome quality, and that unresolved complex repeats and haplotype heterozygosity are major sources of assembly error when not handled correctly. Our assemblies correct substantial errors, add missing sequence in some of the best historical reference genomes, and reveal biological discoveries. These include the identification of many false gene duplications, increases in gene sizes, chromosome rearrangements that are specific to lineages, a repeated independent chromosome breakpoint in bat genomes, and a canonical GC-rich pattern in protein-coding genes and their regulatory regions. Adopting these lessons, we have embarked on the Vertebrate Genomes Project (VGP), an international effort to generate high-quality, complete reference genomes for all of the roughly 70,000 extant vertebrate species and to help to enable a new era of discovery across the life sciences.
Conflict of interest statement
During the contributing period, B.T.H., M. Simbirsky, A.F. and M. Mooney were employees of DNAnexus Inc. S.B.K., R.H., Z.K., J. Korlach, I.S. and C.D. were full-time employees at Pacific Biosciences, a company developing single-molecule long read sequencing technologies. R.E.G., N.H.P., and J.G. were affiliated with Dovetail Genomics, a company developing genome assembly tools, including Hi-C. I.G. was affiliated with Oxford Nanopore Technologies, a company generating long read sequencing technologies. A.H. and J.L were employees of Bionano Genomics, a company developing optical maps for genome assembly. S. Selvaraj was an employee of Arima Genomics, a company developing Hi-C data for genome assemblies. R.D. is a scientific advisory board member of Dovetail Inc. P. Flicek is a member of the Scientific Advisory Boards of Fabric Genomics, Inc., and Eagle Genomics, Ltd. H.C. receives royalties from the sale of UCSC Genome Browser source code, LiftOver, GBiB, and GBiC licenses to commercial entities. S.K. has received travel funds to speak at symposia organized by Oxford Nanopore. M.D. and L.N. receive royalties from licensing of UCSC Genome Browser. For W.E.J., the content here is not to be construed as the views of the DA or DOD. All other authors declare no competing interests.
Figures
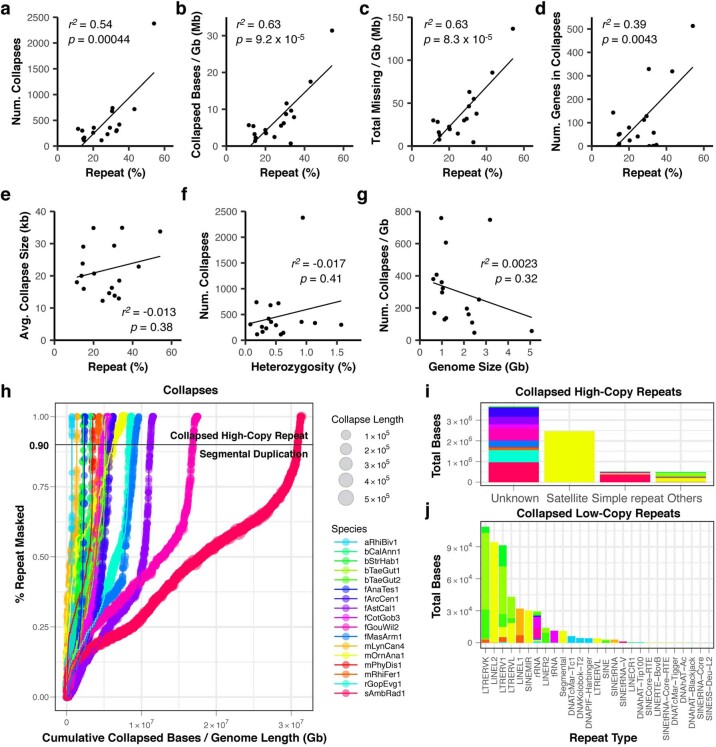
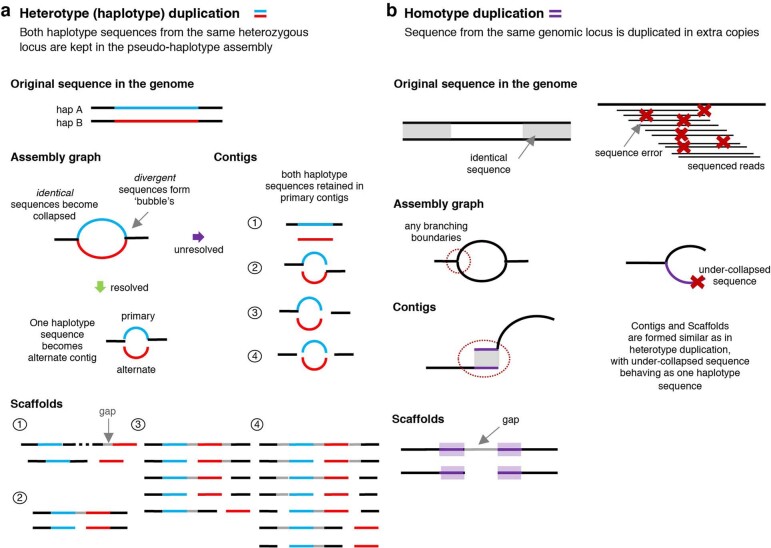

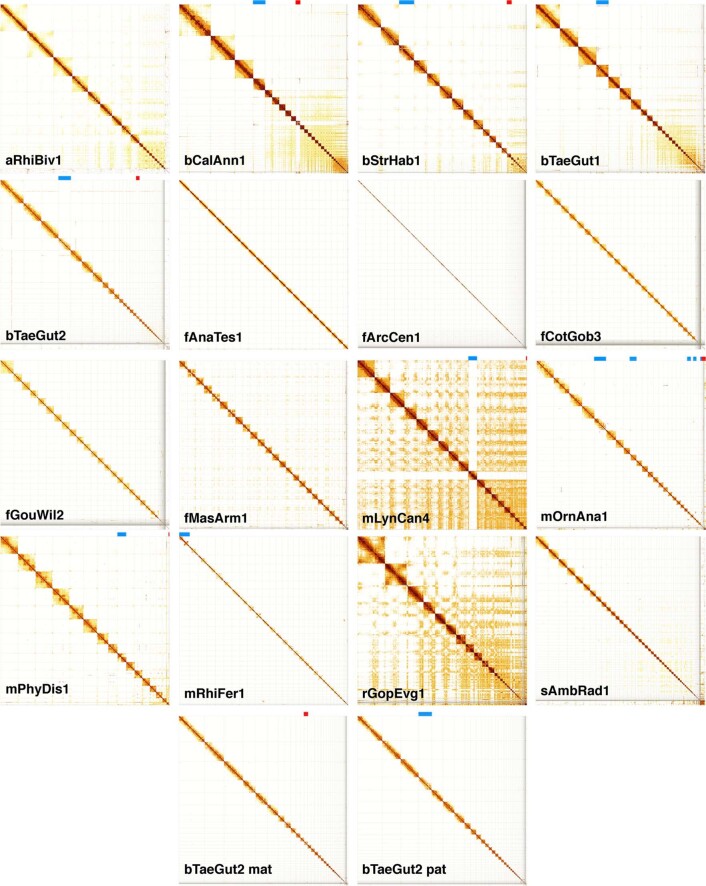
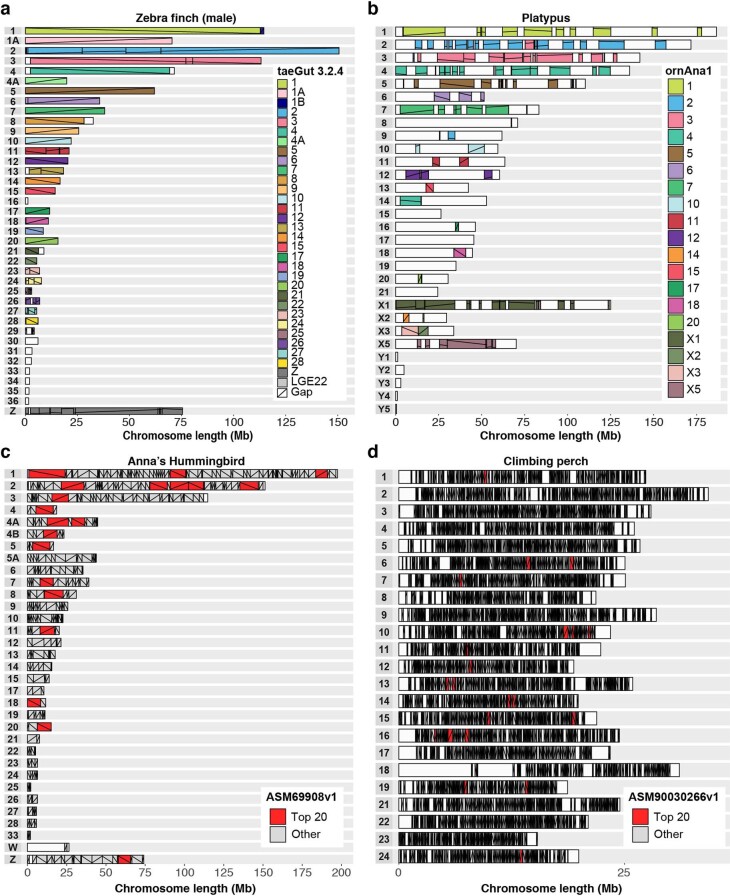
Comment in
-
Assembling vertebrate genomes.Nat Rev Genet. 2021 Jul;22(7):413. doi: 10.1038/s41576-021-00379-z. Nat Rev Genet. 2021. PMID: 34017104 No abstract available.
References
-
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. - PubMed
-
- Sulston J, et al. The C. elegans genome sequencing project: a beginning. Nature. 1992;356:37–41. - PubMed
-
- Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
