

Review
doi: 10.1161/CIRCRESAHA.121.318011.
Epub 2021 Apr 29.
Inflammatory and Biomechanical Drivers of Endothelial-Interstitial Interactions in Calcific Aortic Valve Disease
Affiliations
- PMID: 33914601
- PMCID: PMC8519486
- DOI: 10.1161/CIRCRESAHA.121.318011
Item in Clipboard
Review
Inflammatory and Biomechanical Drivers of Endothelial-Interstitial Interactions in Calcific Aortic Valve Disease
Circ Res.
.
Abstract
Calcific aortic valve disease is dramatically increasing in global burden, yet no therapy exists outside of prosthetic replacement. The increasing proportion of younger and more active patients mandates alternative therapies. Studies suggest a window of opportunity for biologically based diagnostics and therapeutics to alleviate or delay calcific aortic valve disease progression. Advancement, however, has been hampered by limited understanding of the complex mechanisms driving calcific aortic valve disease initiation and progression towards clinically relevant interventions.
Keywords: aortic valve; cardiovascular disease; cytokines; endothelial cells; inflammation.
Figures
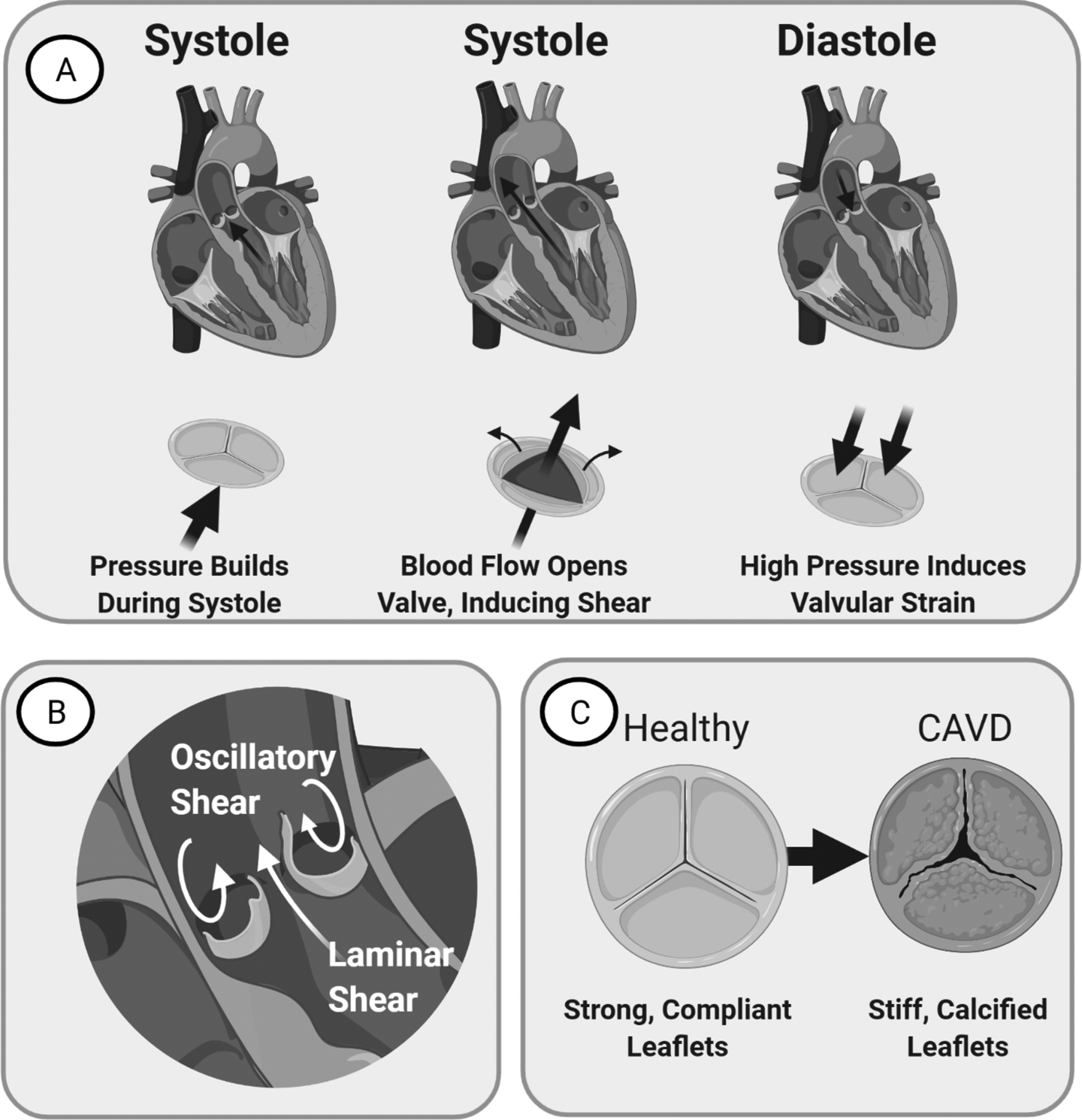
(A) Left: Valve just before systole. Blood flow assists opens the valve. Middle: Valve during Systole. Leaflets ends bend to create an opening for blood flow. Right: Valve during Diastole. Valve provides seal that resists back-flow pressure, inducing strain. (B) Image of the valve demonstrating laminar flow on the ventricularis side and oscillatory flow on the fibrosa side. (C) CAVD manifests as valvular thickening with stiffening and calcification that reduces leaflet mobility over time.
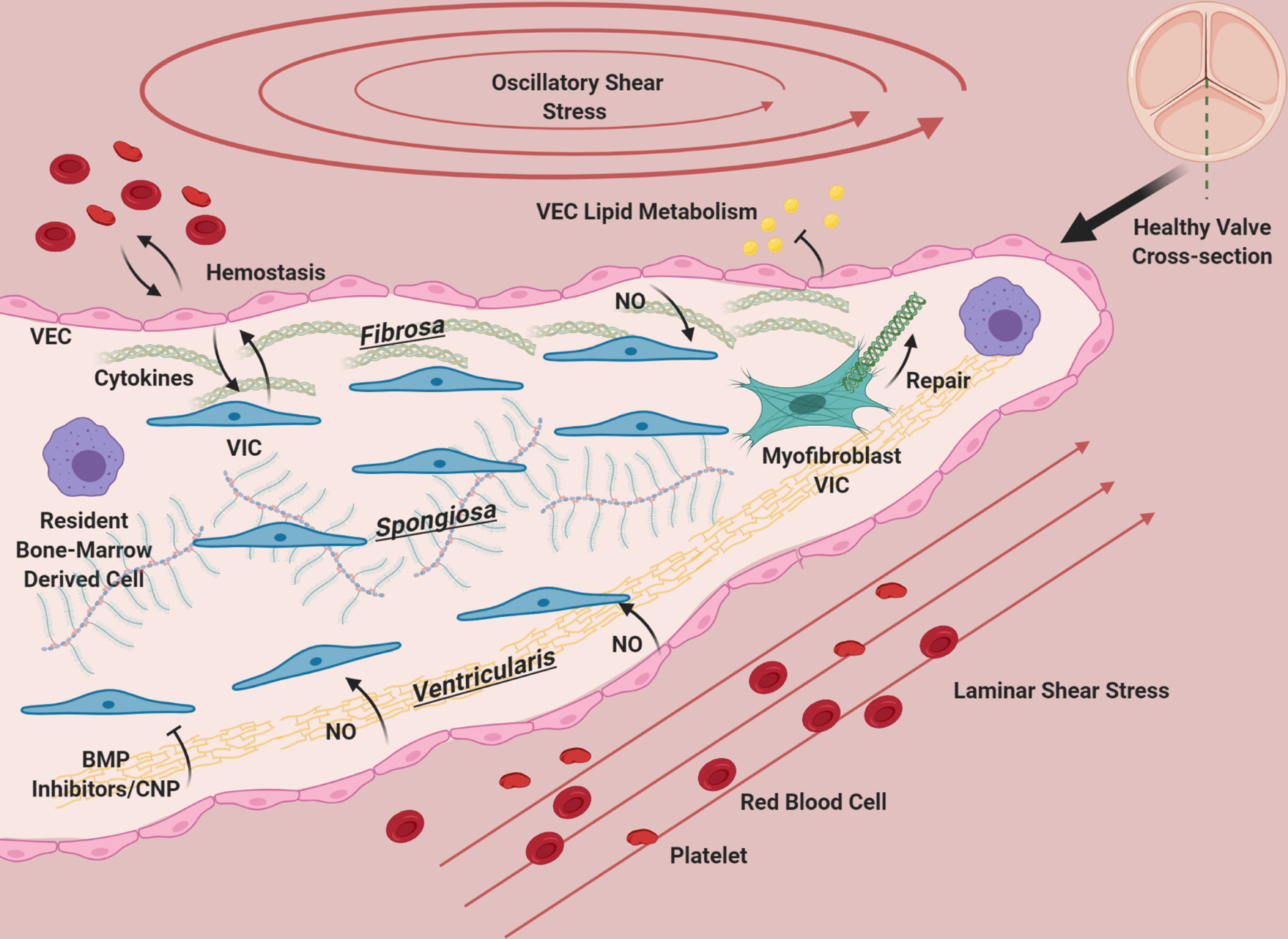
The aortic valve has three layers: the fibrosa, consisting primarily of circumferentially oriented collagen, the spongiosa, consisting primarily of GAGs, and the ventricularis containing radially-aligned elastin. A diverse population of interstitial cells broadly defined as VIC inhabit these layers in the interstitium of the valve (blue) and become activated to remodel the ECM of the valve as needed (green). VEC line the valve and provide a barrier from the blood, preventing clotting, mediating infiltration of lipids, nutrients, and modulating extravasation of inflammatory cells. VEC (pink) promote quiescence in VIC through nitric oxide signaling, and VIC and VEC also communicate with cytokines. Osteogenic inhibitors such as BMP inhibitors and CNP from the ventricularis endothelium likely maintain homeostasis by preventing VIC activation. The homeostatic valve also contains resident immune cells (purple), whose functions are just beginning to be uncovered. The ventricularis of the valve experiences laminar shear, while the fibrosa has oscillatory flow. The valve is constantly under tension/compression as the valve opens and closes, and resists pressure. The ECM composition of the valve and cellular maintenance ensures that it remains flexible, strong and compliant so that it is durable over a human lifespan.
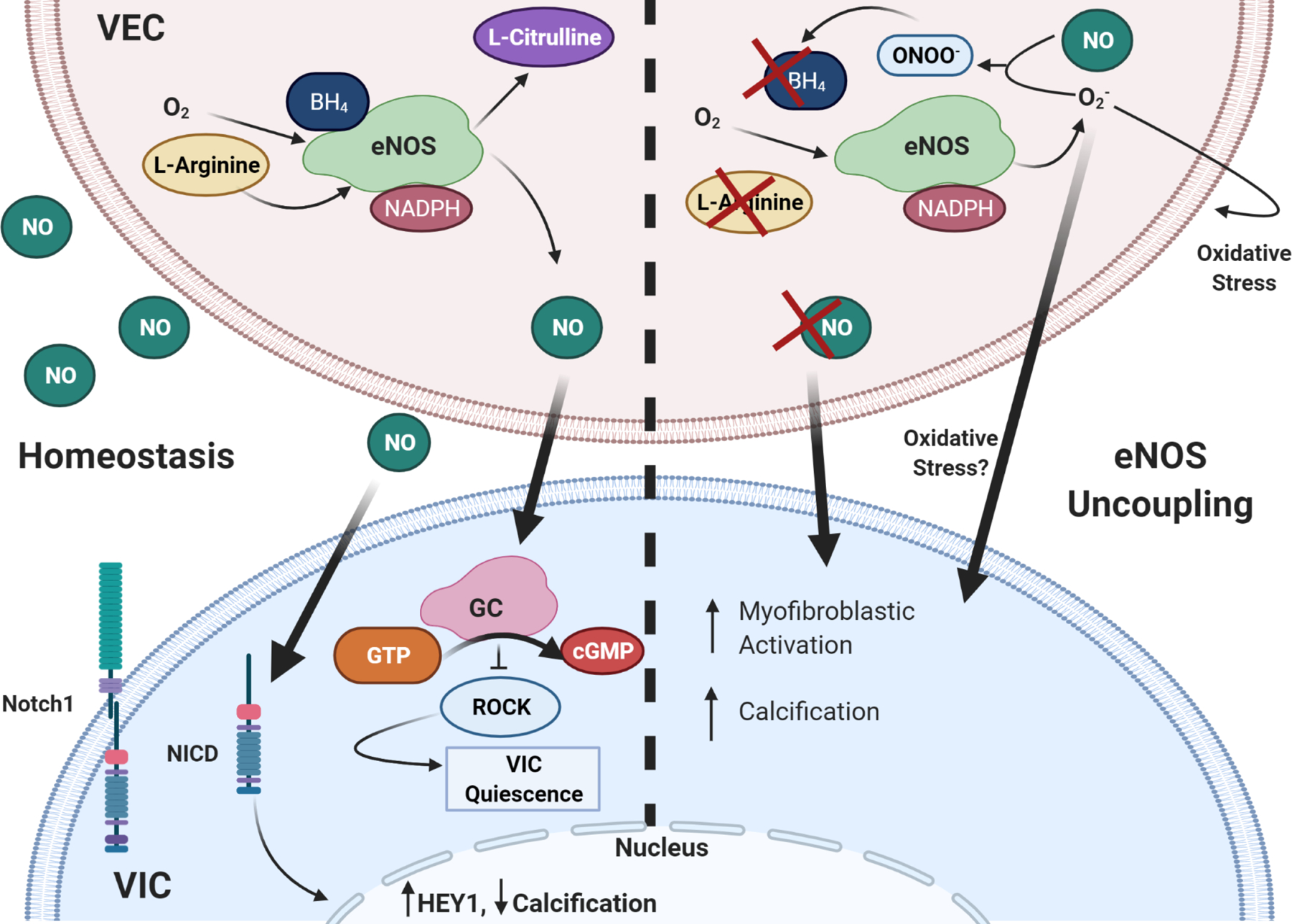
NO signaling from VEC stimulates VIC quiescence through activation of GC/cGMP pathway and subsequent downregulation of RhoA pathway. It also prevents calcification through assisting with Notch1 translocation to the nucleus. eNOS uncoupling causes endothelial dysfunction, such that there is little to no NO signaling to VIC, increasing myofibroblastic activation and calcification. ROS is known to induce cell damage, Superoxide can further cause oxidative stress in VEC and VIC. Thick arrows are VIC-VEC signaling, thin are intracellular or autocrine VEC effects.
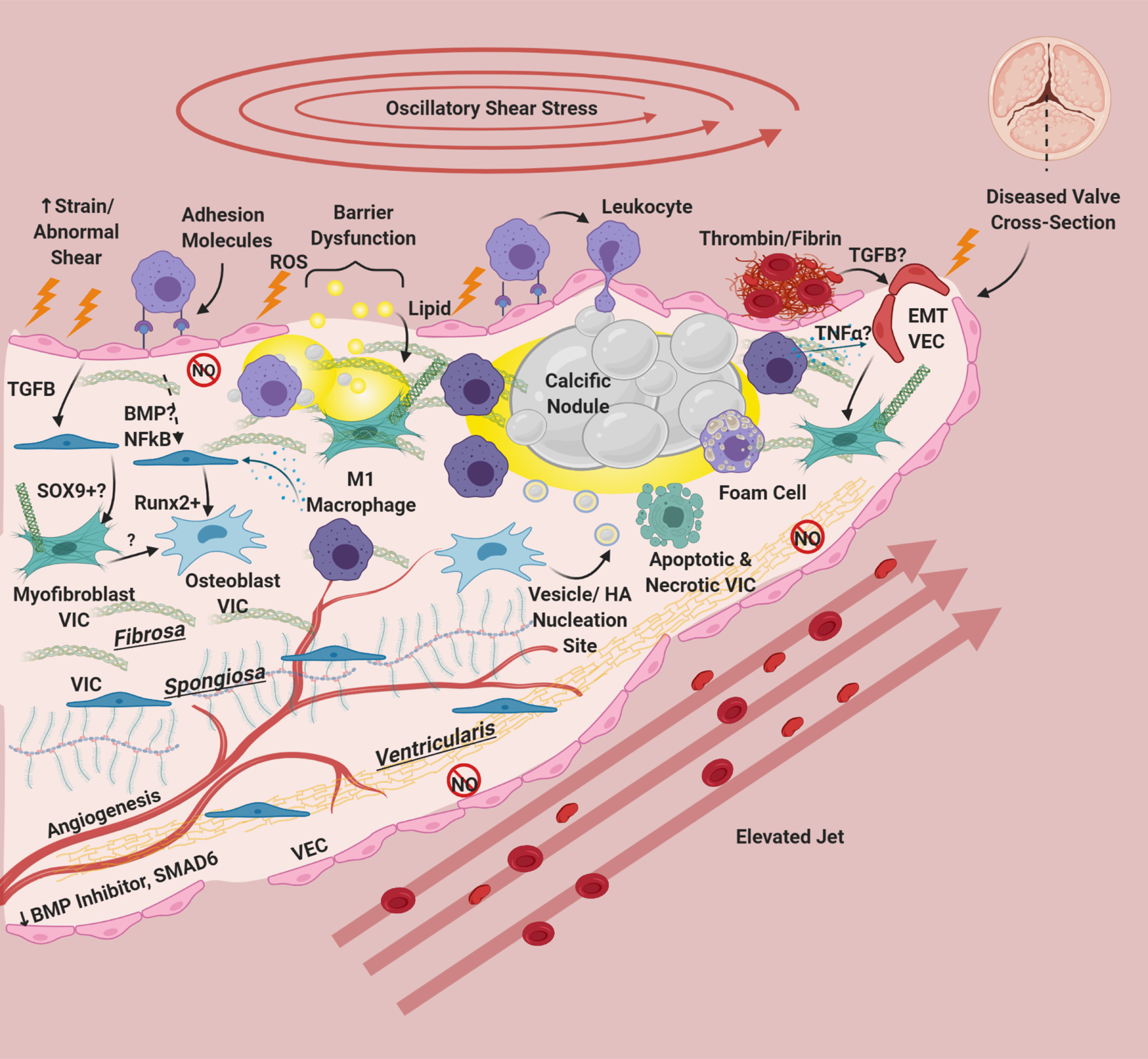
Risk factors cause damage, inflammatory adhesion expression, barrier disruption, and lipid deposition in/underneath the endothelium. Deposited lipids, specifically oxidized lipids can cause inflammation and damage in resident cells. Endothelial dysfunction results in eNOS down-regulation and dysfunction, abrogating protective eNOS signaling to VIC and in hemostasis. This is especially apparent on the ventricularis side of the valve. There is also a reduction in protective BMP inhibitors and SMAD6 on the ventricularis side of the valve. Leukocytes bind to VEC adhesion molecules and undergo extravasation into the tissue. Clotting factors including thrombin, tissue factor, and fibrin have been associated with calcified valves, and platelets recently have been implicated as sources of TGFB in valve disease. TGFB from VEC has been shown to induce Sox9 expression and to produce activated myofibroblast-like VIC. Paracrine signals from macrophages, specifically M1 macrophages has been shown to induce osteogenic differentiation of VIC. NFκB and BMP (possibly) from VEC can also modulate VIC osteoblastic differentiation. Osteoblastic VIC may excrete vesicles or undergo apoptosis/necrosis that deposit calcium and create HA nucleation sites, which grow over time to form large calcific nodules. Inflammatory infiltrate localizes around nodules, and areas of angiogenesis. Foam cells are observed in CAVD, but do not form dense necrotic cores. Inflammatory cytokines like TGFB and TNFα can cause VEC to undergo EndMT, creating fibrotic activated VIC-like cells. Thickening of the valve happens not only through lipid deposition/calcification but also fibrotic remodeling from activated VIC. CAVD induces angiogenesis in the normally avascular leaflet. For reasons unknown but hypothesized to be due to laminar shear stress, the ventricularis of the CAVD valve remains largely unaffected
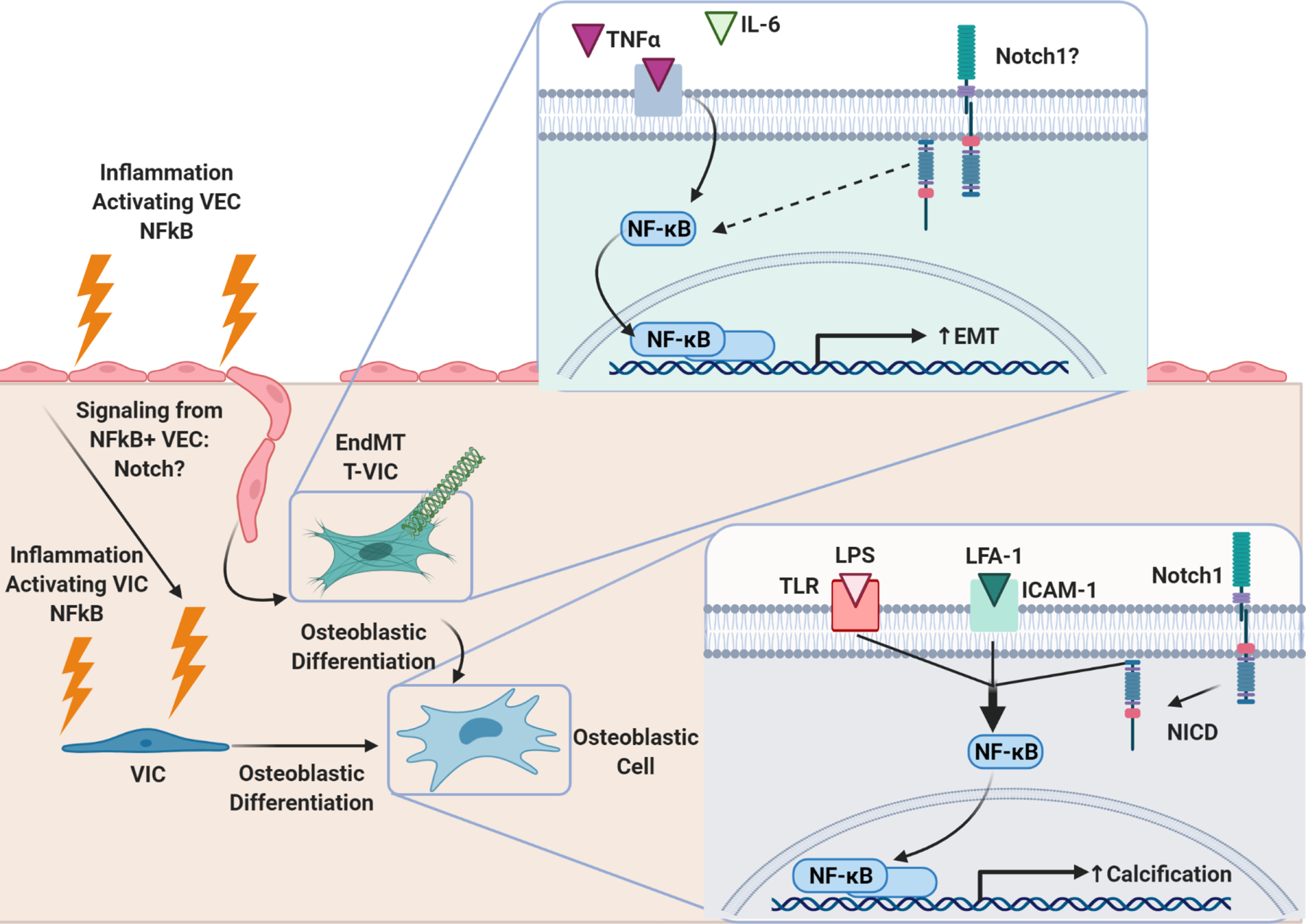
Bottom Right: NFκB activation in VIC through concerted efforts of inflammatory LPS/TLR signaling, LFA-1/ICAM-1 signaling, and Notch-1 signaling induces VIC osteoblastic differentiation and calcification. Left: Inflammation induces NFκB activation in VEC. NFκB+ VEC undergo EndMT, transforming into VIC (T-VIC). NFκB+ VEC have also been shown to exhibit evidence of osteoblastic differentiation. Knock-out of valve-specific NFκB in CAVD models prevents calcification and adverse valvular remodeling, consistent with the paradigm that NFκB activation in VEC promotes pathologic differentiation of VIC. Top right: Inflammatory cytokines TNFα and IL-6 activate NFκB signaling in VEC, which induces EndMT. It is unknown if Notch1 also plays a role in NFκB signaling in VEC. If so, Notch pathway could be a method through which VIC and VEC communicate in response to inflammation.
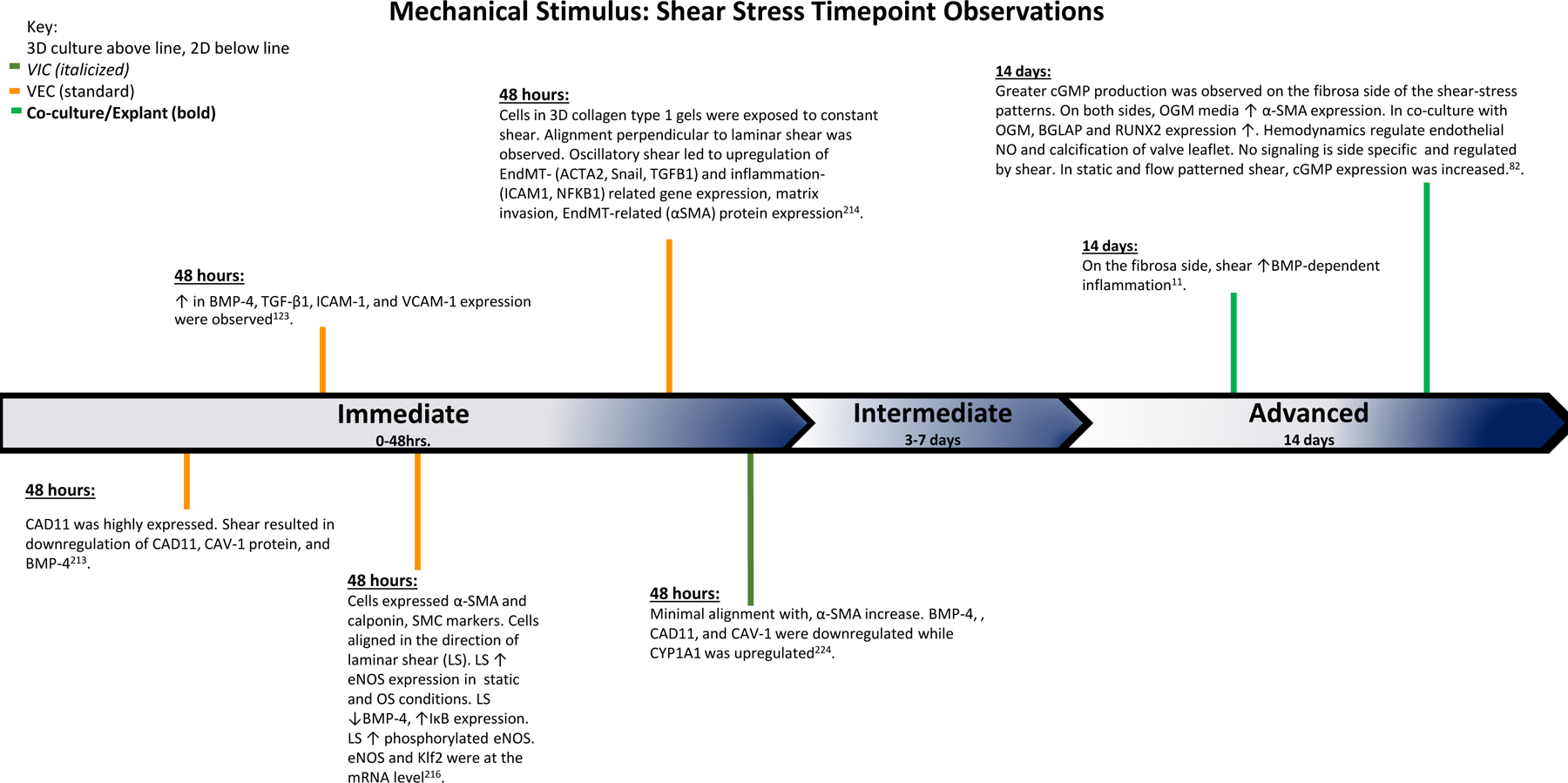
The findings of various studies observing the effects of shear stress over the course of the experiments. Based on the time points of experimental findings, the observations are classified as Immediate (0–48 hrs.), Intermediate (3–7 days), and Advanced (14 days).
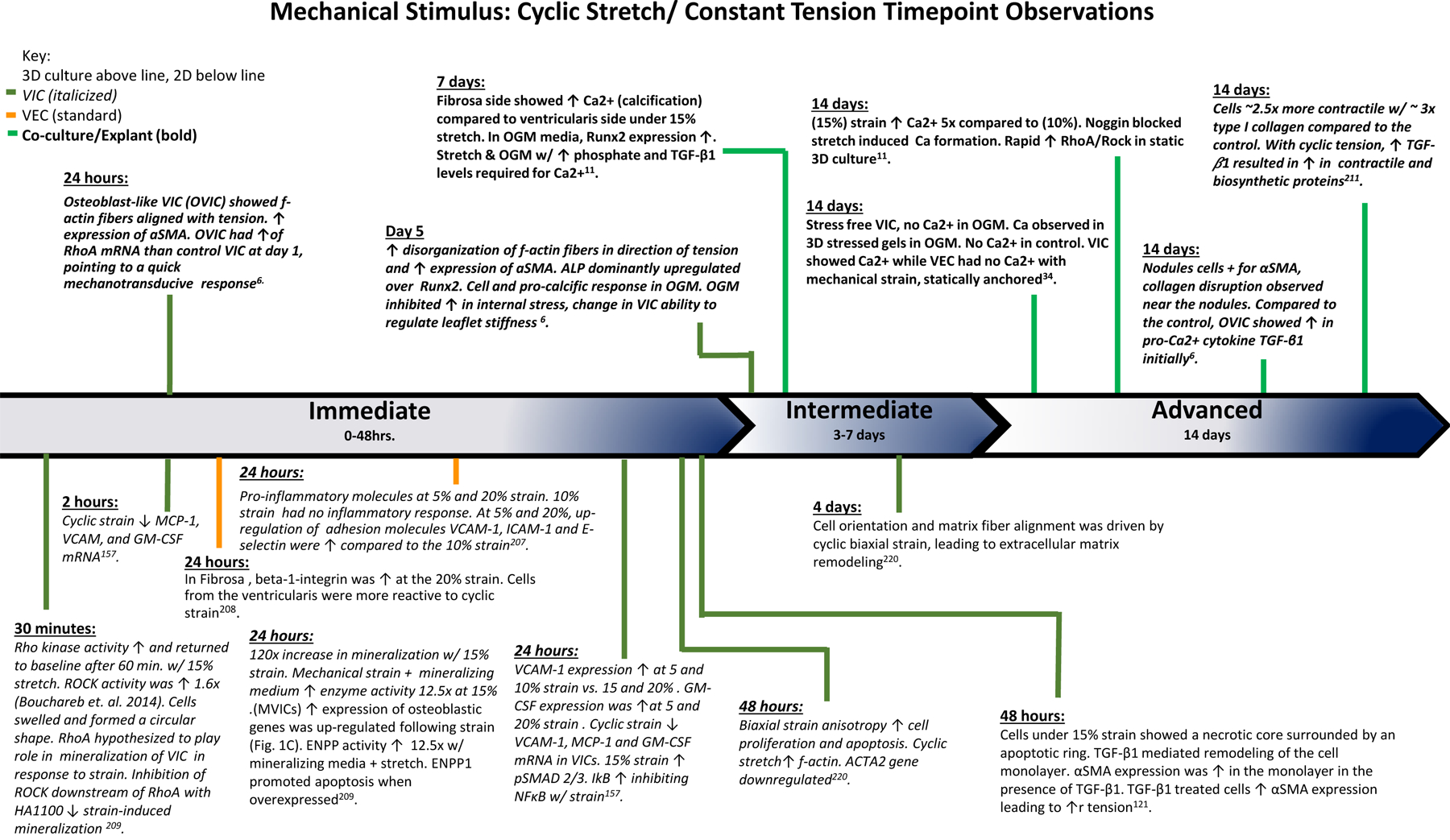
The findings of various studies observing the effects of cyclic stretch and constant tension over the course of the experiments. Based on the time points of experimental findings, the observations are classified as Immediate (0–48 hrs.), Intermediate (3–7 days), and Advanced (14 days).
References
-
- Thubrikar M, Bosher LP, Nolan SP. The mechanism of opening of the aortic valve. J Thorac Cardiovasc Surg 1979;77:863–870. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
