

Prophages integrating into prophages: A mechanism to accumulate type III secretion effector genes and duplicate Shiga toxin-encoding prophages in Escherichia coli
- PMID: 33914852
- PMCID: PMC8112680
- DOI: 10.1371/journal.ppat.1009073
Prophages integrating into prophages: A mechanism to accumulate type III secretion effector genes and duplicate Shiga toxin-encoding prophages in Escherichia coli
Abstract
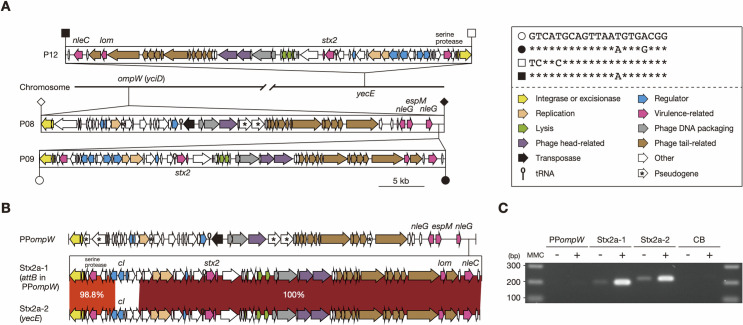
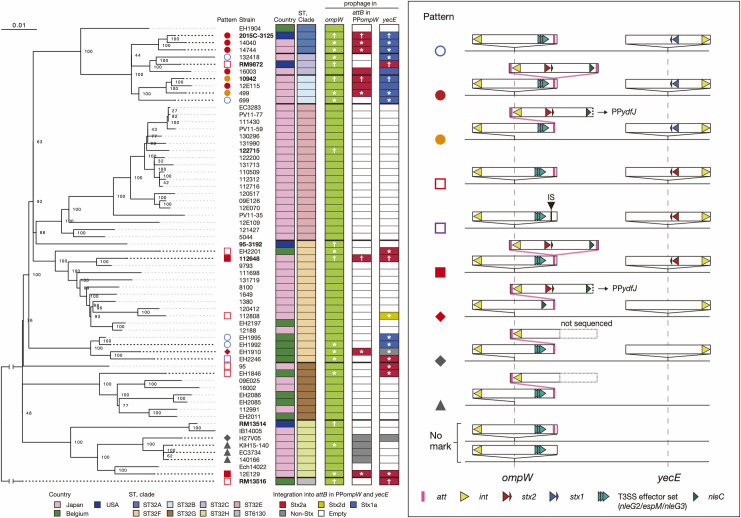
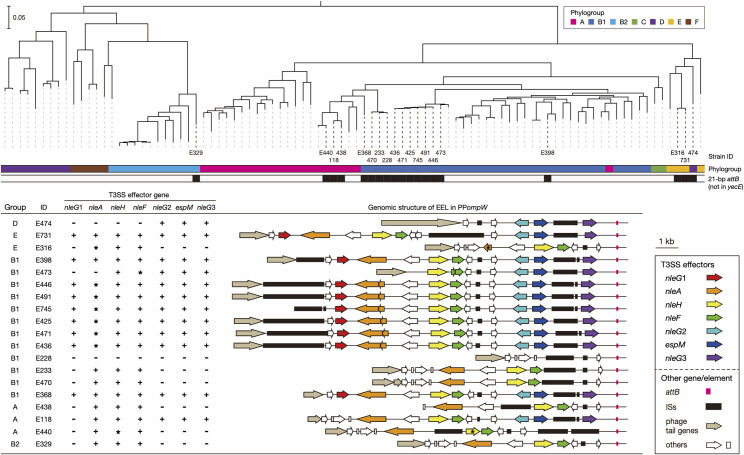
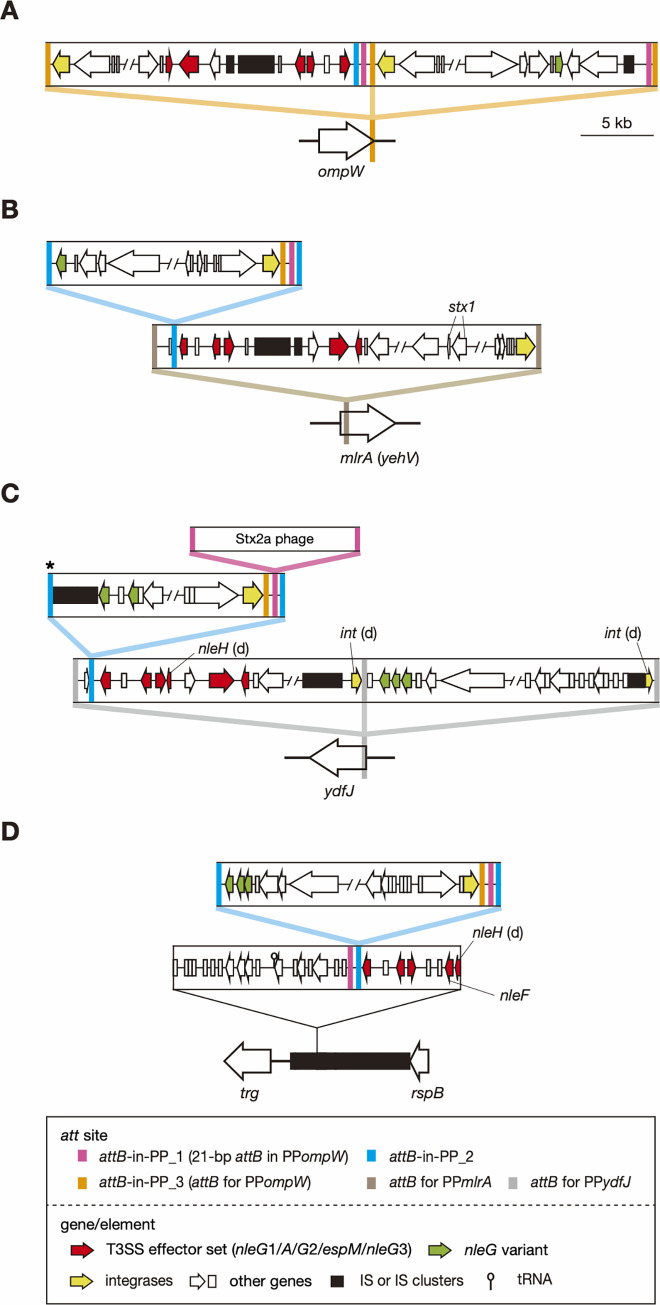
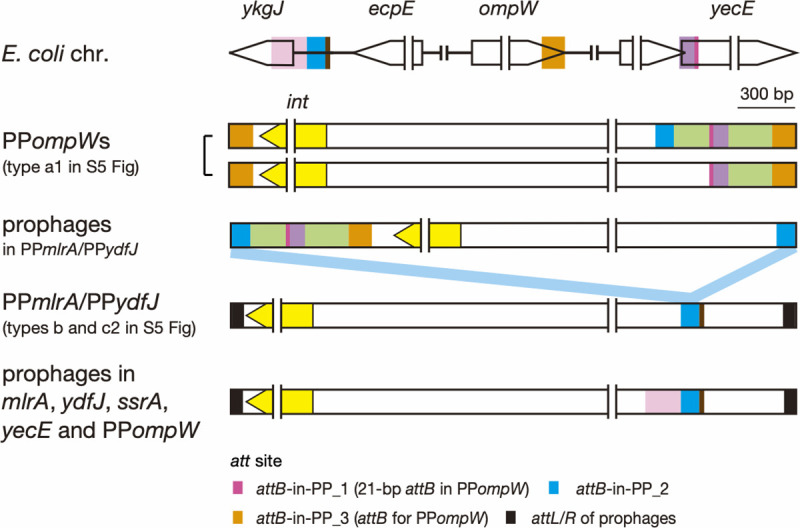
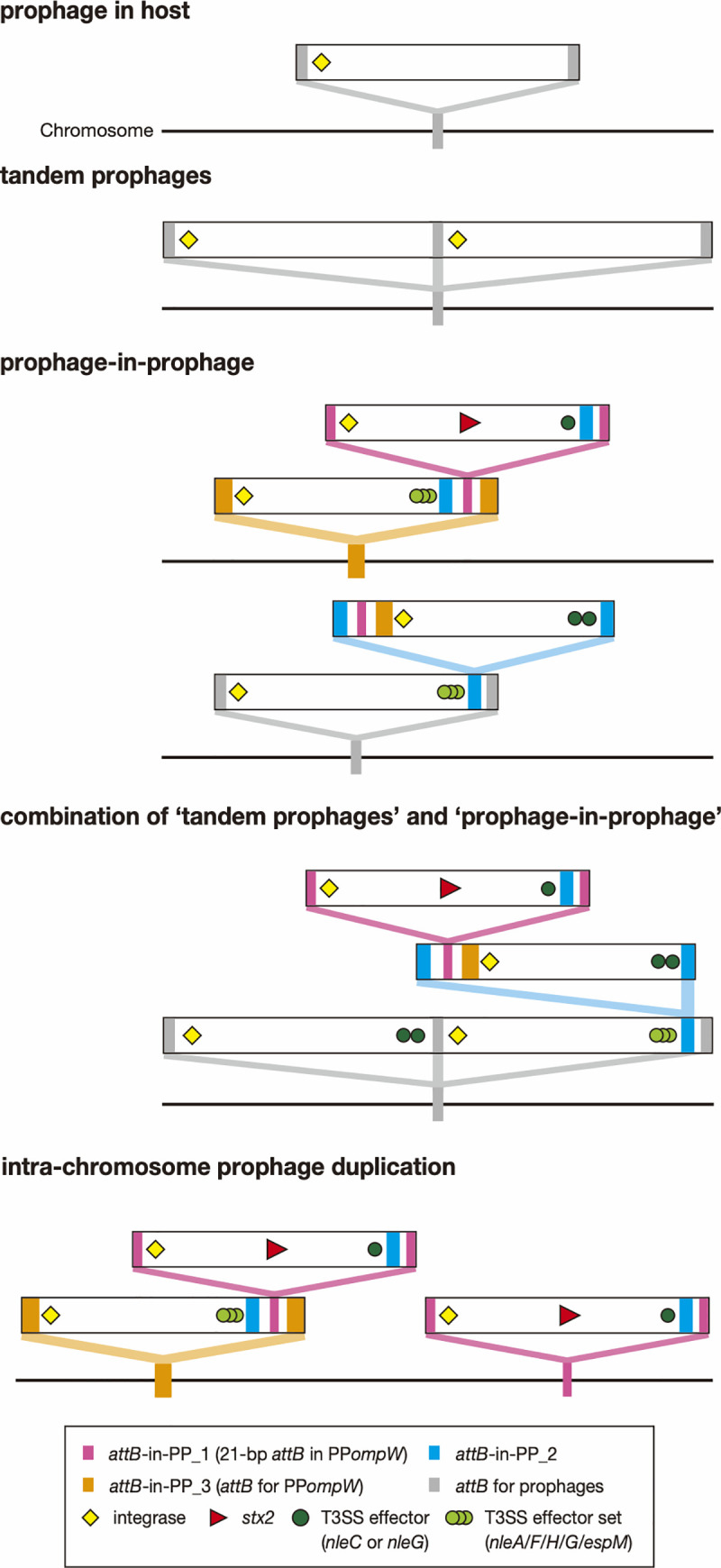
Bacteriophages (or phages) play major roles in the evolution of bacterial pathogens via horizontal gene transfer. Multiple phages are often integrated in a host chromosome as prophages, not only carrying various novel virulence-related genetic determinants into host bacteria but also providing various possibilities for prophage-prophage interactions in bacterial cells. In particular, Escherichia coli strains such as Shiga toxin (Stx)-producing E. coli (STEC) and enteropathogenic E. coli (EPEC) strains have acquired more than 10 prophages (up to 21 prophages), many of which encode type III secretion system (T3SS) effector gene clusters. In these strains, some prophages are present at a single locus in tandem, which is usually interpreted as the integration of phages that use the same attachment (att) sequence. Here, we present phages integrating into T3SS effector gene cluster-associated loci in prophages, which are widely distributed in STEC and EPEC. Some of the phages integrated into prophages are Stx-encoding phages (Stx phages) and have induced the duplication of Stx phages in a single cell. The identified attB sequences in prophage genomes are apparently derived from host chromosomes. In addition, two or three different attB sequences are present in some prophages, which results in the generation of prophage clusters in various complex configurations. These phages integrating into prophages represent a medically and biologically important type of inter-phage interaction that promotes the accumulation of T3SS effector genes in STEC and EPEC, the duplication of Stx phages in STEC, and the conversion of EPEC to STEC and that may be distributed in other types of E. coli strains as well as other prophage-rich bacterial species.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Differential induction of Shiga toxin in environmental Escherichia coli O145:H28 strains carrying the same genotype as the outbreak strains.Int J Food Microbiol. 2021 Feb 2;339:109029. doi: 10.1016/j.ijfoodmicro.2020.109029. Epub 2020 Dec 23. Int J Food Microbiol. 2021. PMID: 33360585
-
Shiga toxin-producing Escherichia coli strains from cattle as a source of the Stx2a bacteriophages present in enteroaggregative Escherichia coli O104:H4 strains.Int J Med Microbiol. 2013 Dec;303(8):595-602. doi: 10.1016/j.ijmm.2013.08.001. Epub 2013 Aug 16. Int J Med Microbiol. 2013. PMID: 24012149
-
The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants.PLoS Pathog. 2009 May;5(5):e1000408. doi: 10.1371/journal.ppat.1000408. Epub 2009 May 1. PLoS Pathog. 2009. PMID: 19412337 Free PMC article.
-
Bacteriophages of Shiga Toxin-Producing Escherichia coli and Their Contribution to Pathogenicity.Pathogens. 2021 Mar 29;10(4):404. doi: 10.3390/pathogens10040404. Pathogens. 2021. PMID: 33805526 Free PMC article. Review.
-
Implications of free Shiga toxin-converting bacteriophages occurring outside bacteria for the evolution and the detection of Shiga toxin-producing Escherichia coli.Front Cell Infect Microbiol. 2014 Apr 16;4:46. doi: 10.3389/fcimb.2014.00046. eCollection 2014. Front Cell Infect Microbiol. 2014. PMID: 24795866 Free PMC article. Review.
Cited by
-
Diversity of Shiga toxin transducing phages in Escherichia coli O145:H28 and the different Shiga toxin 2 production levels associated with short- or long-tailed phages.Front Microbiol. 2024 Aug 6;15:1453887. doi: 10.3389/fmicb.2024.1453887. eCollection 2024. Front Microbiol. 2024. PMID: 39165568 Free PMC article.
-
Streptococcus pneumoniae: a Plethora of Temperate Bacteriophages With a Role in Host Genome Rearrangement.Front Cell Infect Microbiol. 2021 Nov 18;11:775402. doi: 10.3389/fcimb.2021.775402. eCollection 2021. Front Cell Infect Microbiol. 2021. PMID: 34869076 Free PMC article.
-
The Prophage and Us-Shiga Toxin Phages Revisited.Pathogens. 2023 Feb 2;12(2):232. doi: 10.3390/pathogens12020232. Pathogens. 2023. PMID: 36839504 Free PMC article.
-
A prophage encoded ribosomal RNA methyltransferase regulates the virulence of Shiga-toxin-producing Escherichia coli (STEC).Nucleic Acids Res. 2024 Jan 25;52(2):856-871. doi: 10.1093/nar/gkad1150. Nucleic Acids Res. 2024. PMID: 38084890 Free PMC article.
-
Combined usage of serodiagnosis and O antigen typing to isolate Shiga toxin-producing Escherichia coli O76:H7 from a hemolytic uremic syndrome case and genomic insights from the isolate.Microbiol Spectr. 2024 Jan 11;12(1):e0235523. doi: 10.1128/spectrum.02355-23. Epub 2023 Dec 4. Microbiol Spectr. 2024. PMID: 38092668 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
