

Calculating Sample Size Requirements for Temporal Dynamics in Single-Cell Proteomics
- PMID: 33915259
- PMCID: PMC8165548
- DOI: 10.1016/j.mcpro.2021.100085
Calculating Sample Size Requirements for Temporal Dynamics in Single-Cell Proteomics
Abstract
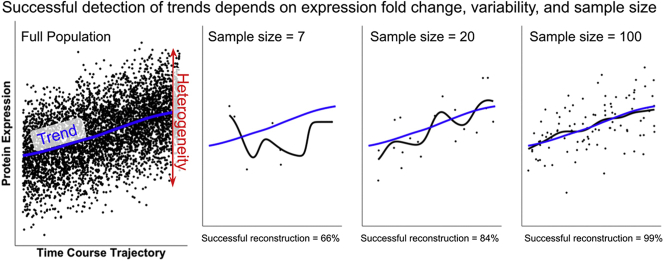
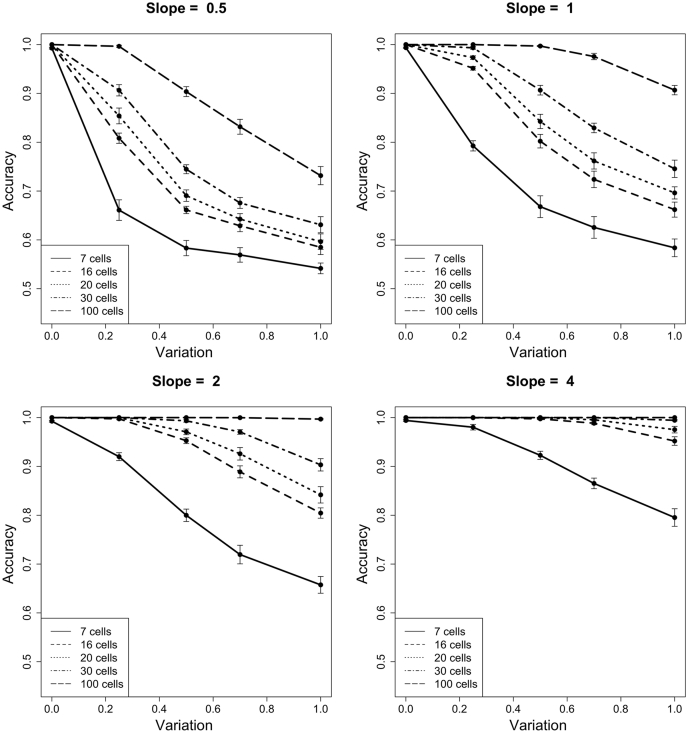
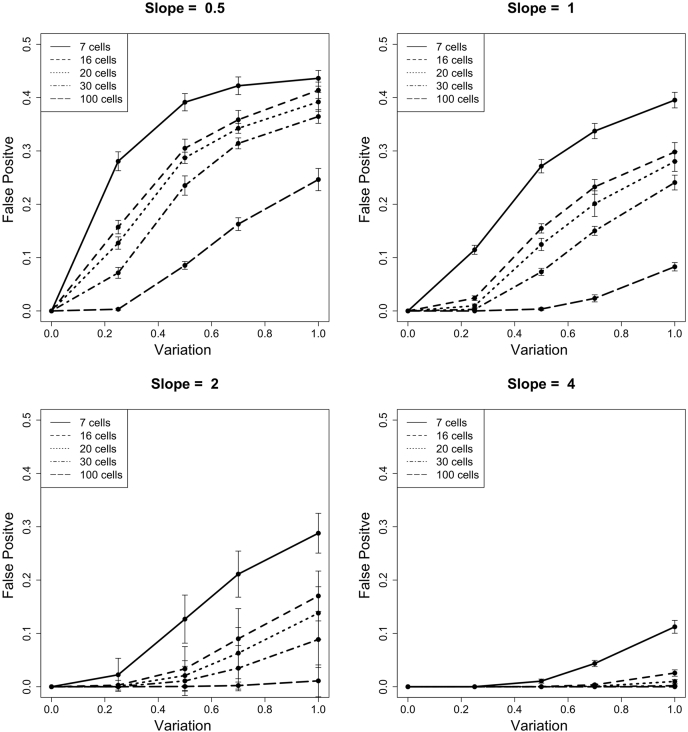
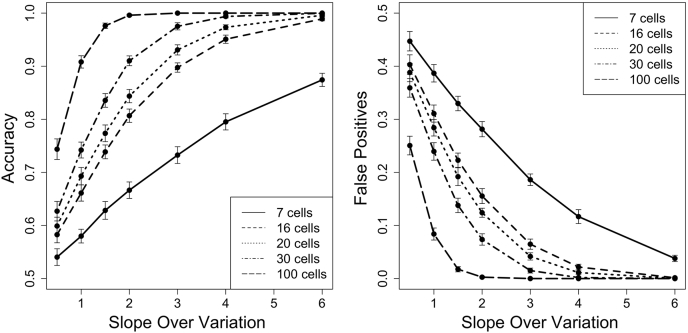
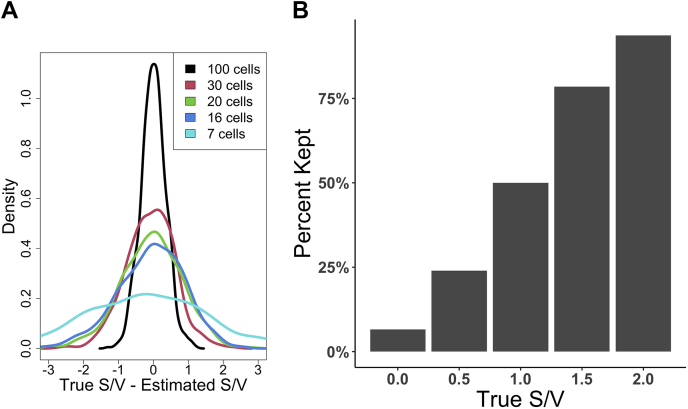
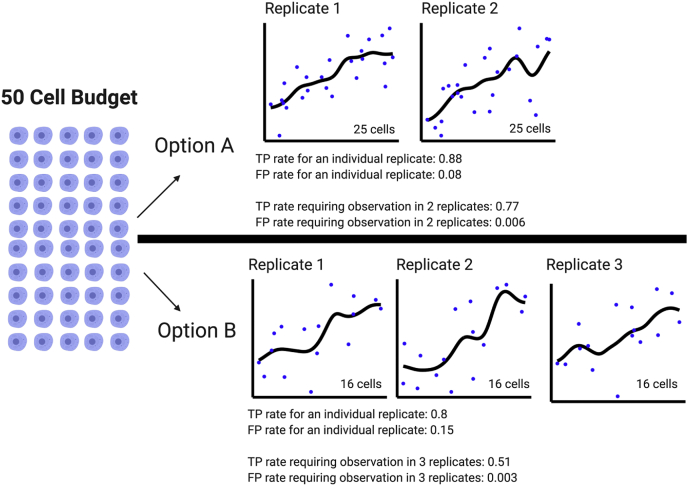
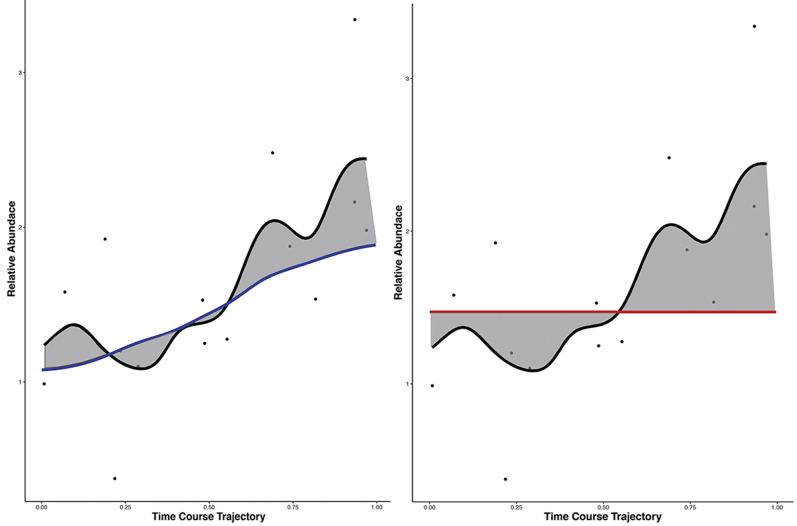
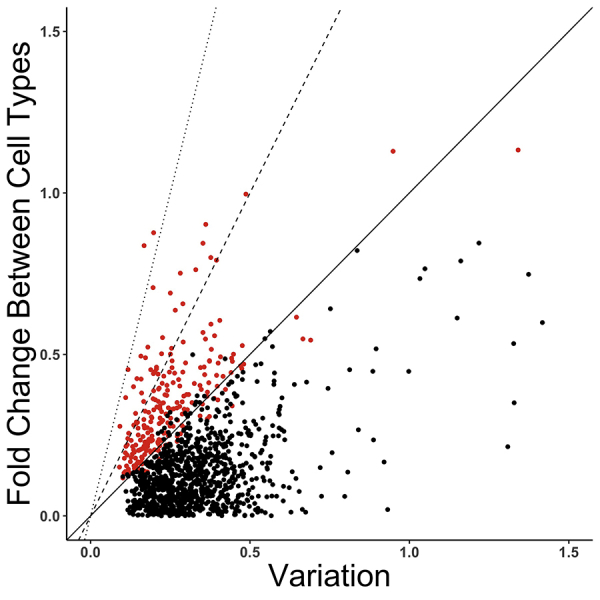
Single-cell measurements are uniquely capable of characterizing cell-to-cell heterogeneity and have been used to explore the large diversity of cell types and physiological functions present in tissues and other complex cell assemblies. An intriguing application of single-cell proteomics is the characterization of proteome dynamics during biological transitions, like cellular differentiation or disease progression. Time-course experiments, which regularly take measurements during state transitions, rely on the ability to detect dynamic trajectories in a data series. However, in a single-cell proteomics experiment, cell-to-cell heterogeneity complicates the confident identification of proteome dynamics as measurement variability may be higher than expected. Therefore, a critical question for these experiments is how many data points need to be acquired during the time course to enable robust statistical analysis. We present here an analysis of the most important variables that affect statistical confidence in the detection of proteome dynamics: fold change, measurement variability, and the number of cells measured during the time course. Importantly, we show that datasets with less than 16 measurements across the time domain suffer from low accuracy and also have a high false-positive rate. We also demonstrate how to balance competing demands in experimental design to achieve a desired result.
Keywords: bioinformatics; experimental design; single-cell proteomics.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare no competing interests.
Figures
References
-
- Mahdessian D., Cesnik A.J., Gnann C., Danielsson F., Stenström L., Arif M., Zhang C., Le T., Johansson F., Shutten R., Bäckström A., Axelsson U., Thul P., Cho N.H., Carja O. Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature. 2021;590:649–654. - PubMed
-
- Doerr A. Single-cell proteomics. Nat. Methods. 2019;16:20. - PubMed
-
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., Trombetta J.J., Weitz D.A., Sanes J.R., Shalek A.K., Regev A. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
