

COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets
- PMID: 33915569
- PMCID: PMC8919505
- DOI: 10.1038/s41586-021-03570-8
COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets
Abstract
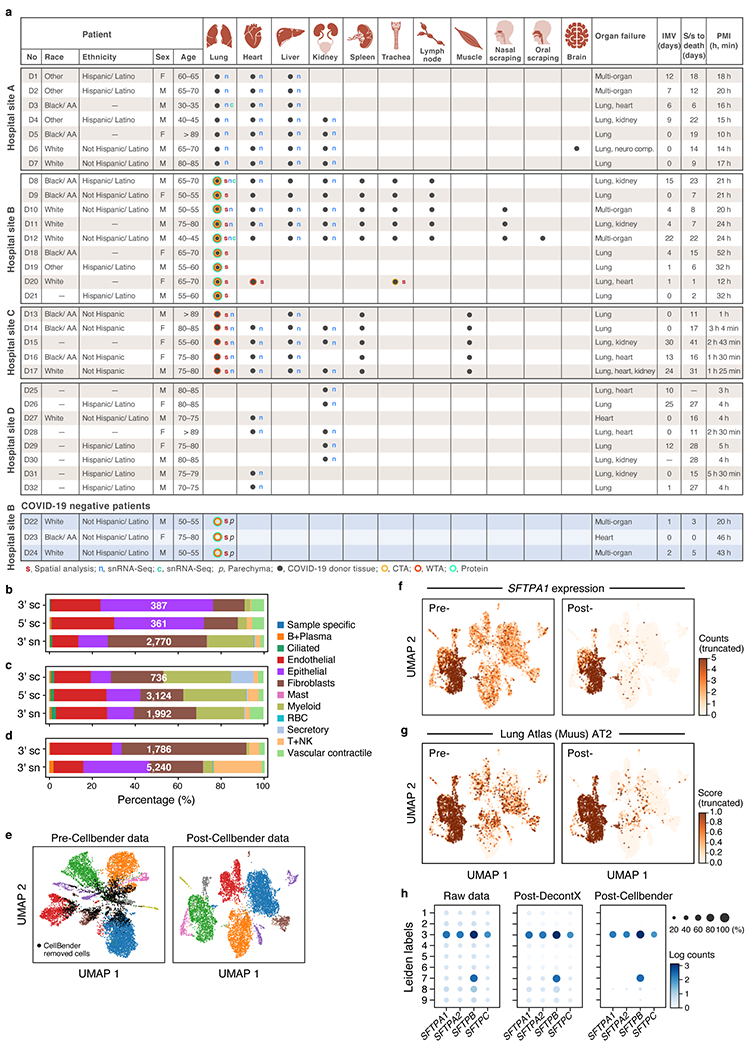
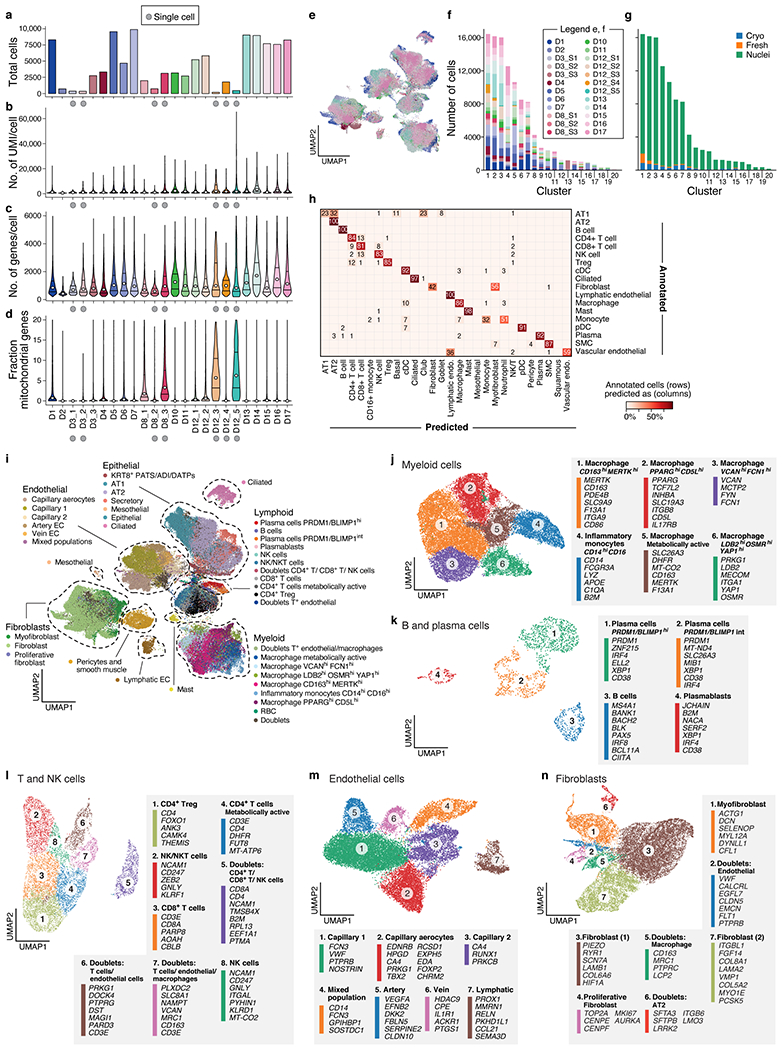
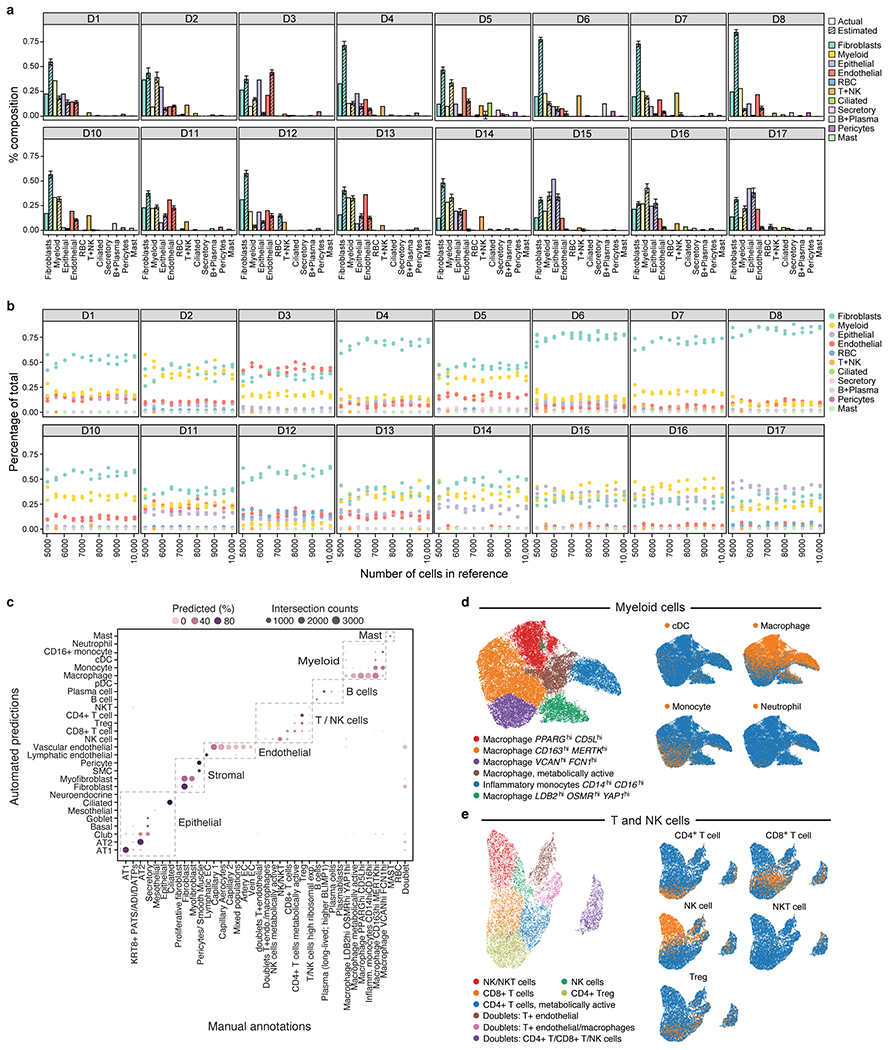
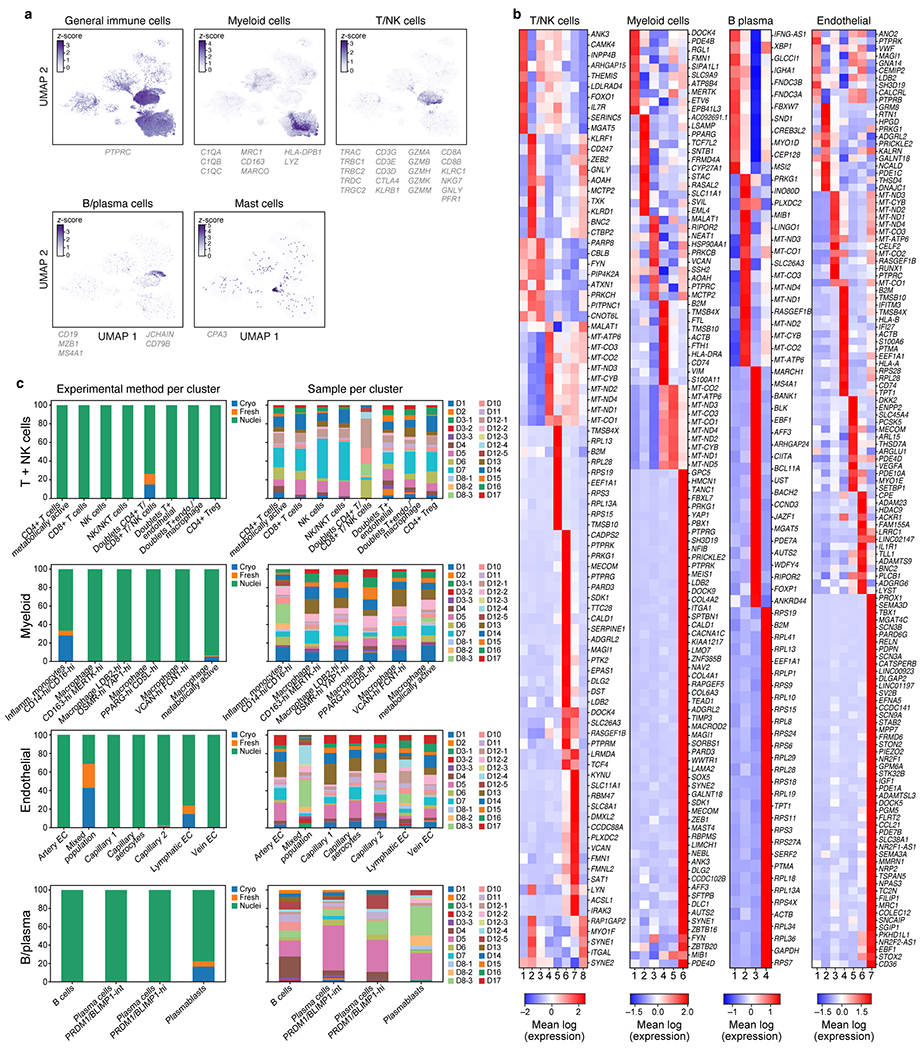
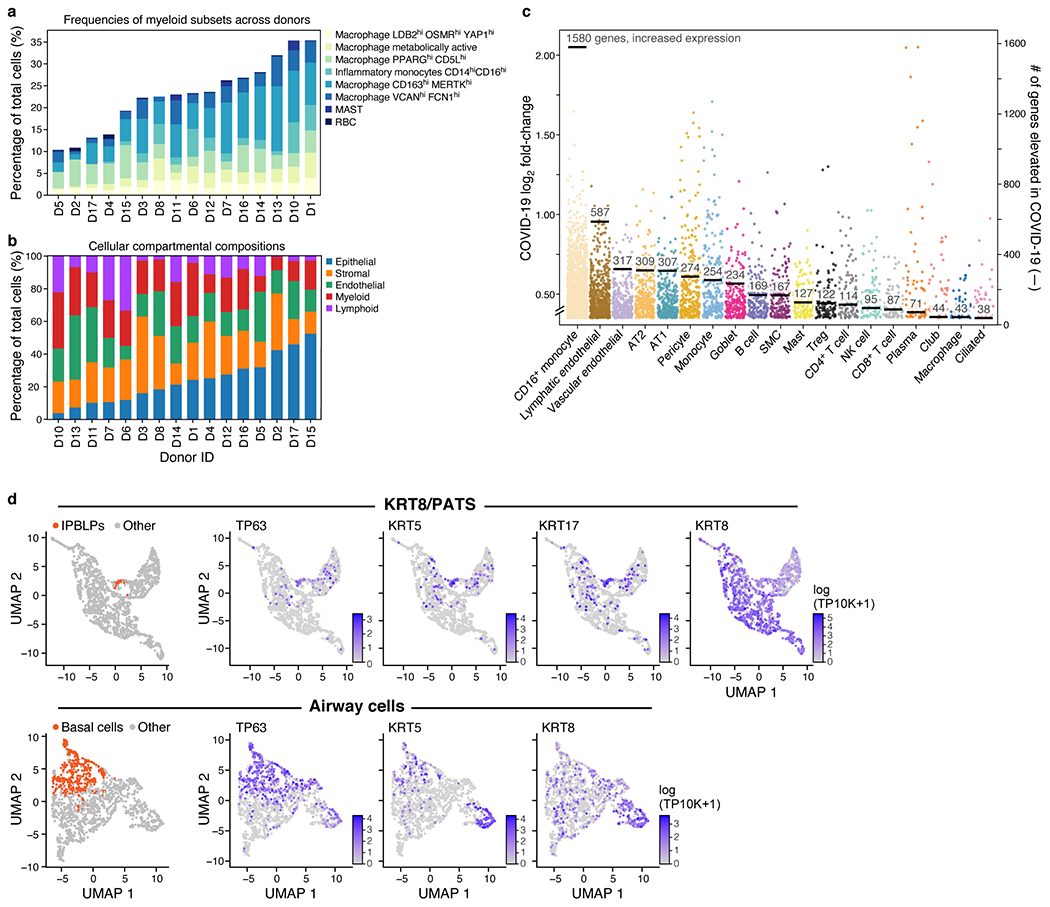
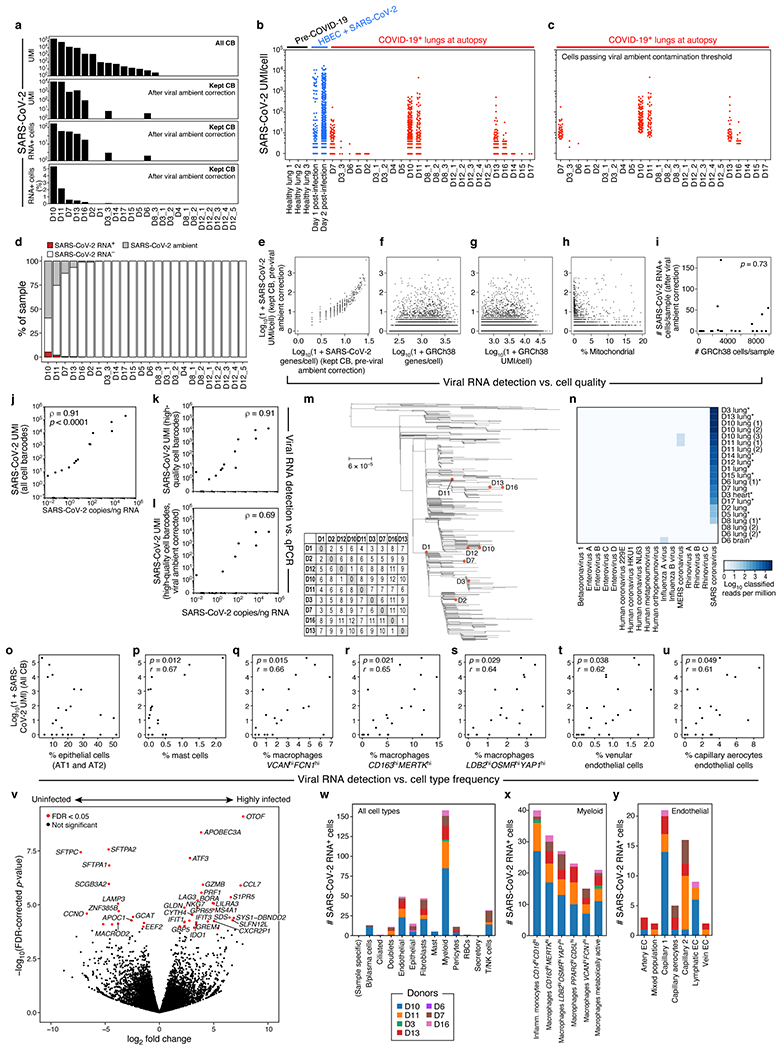
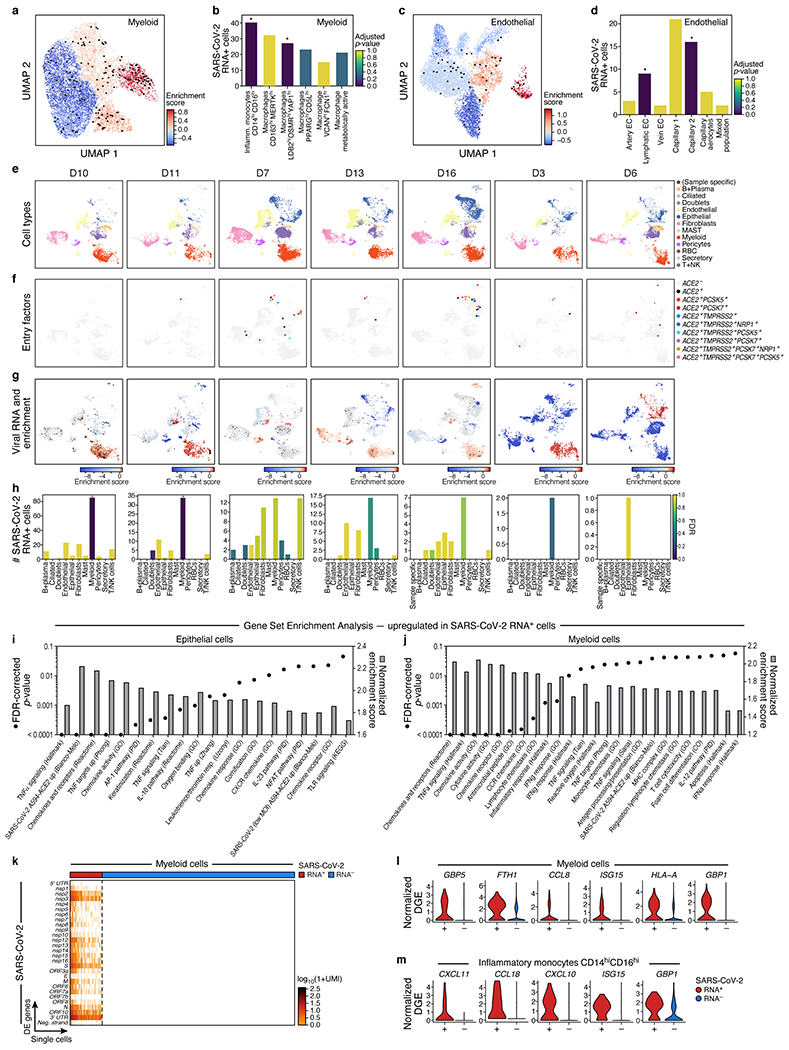
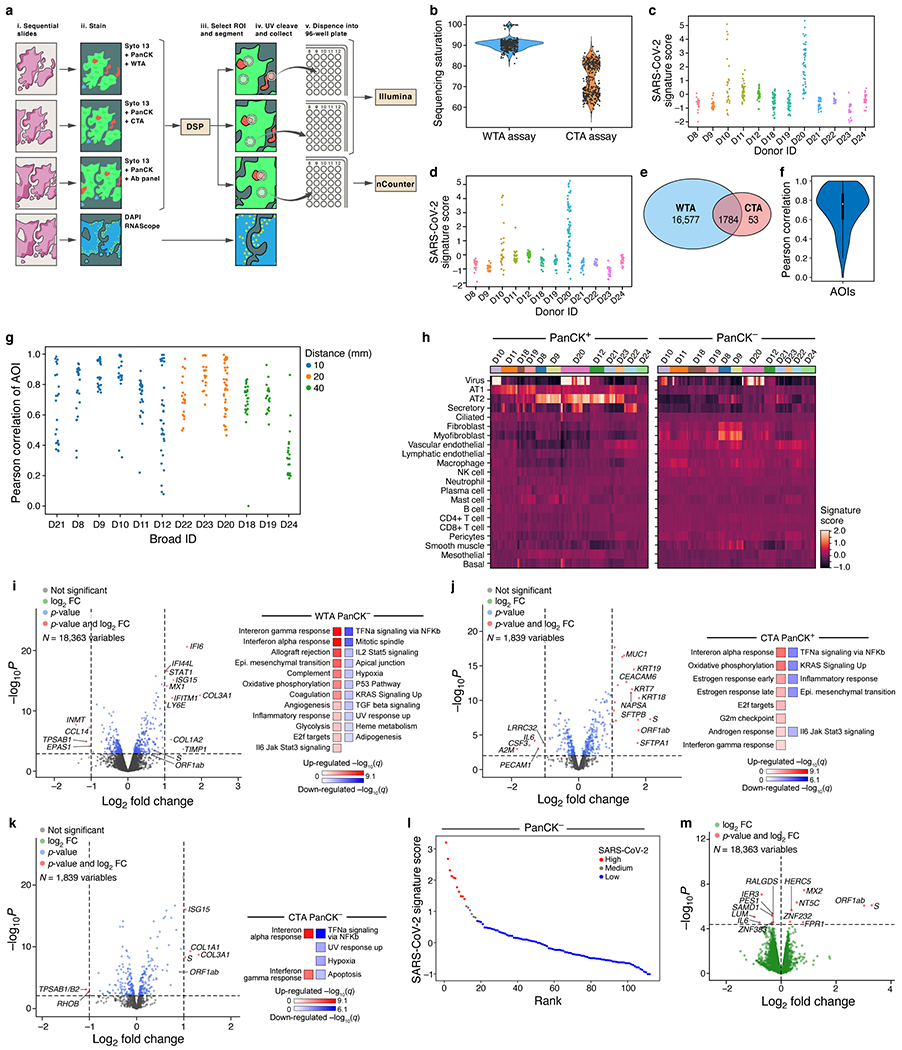
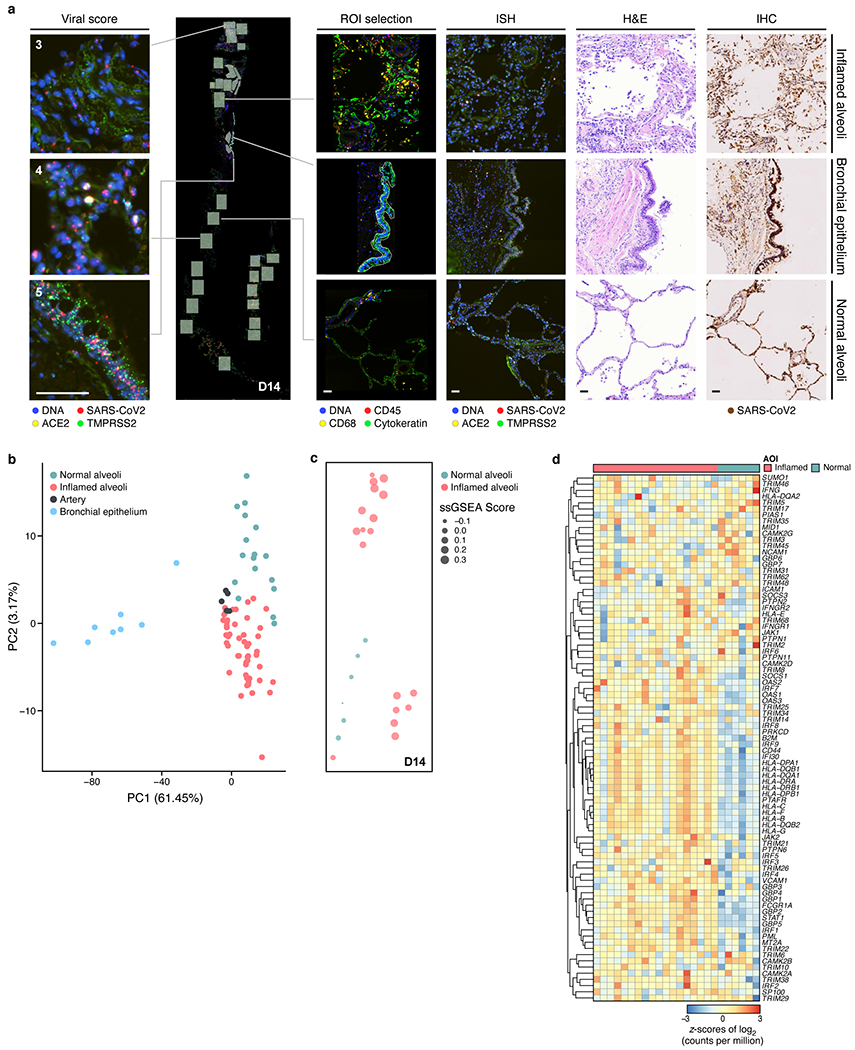
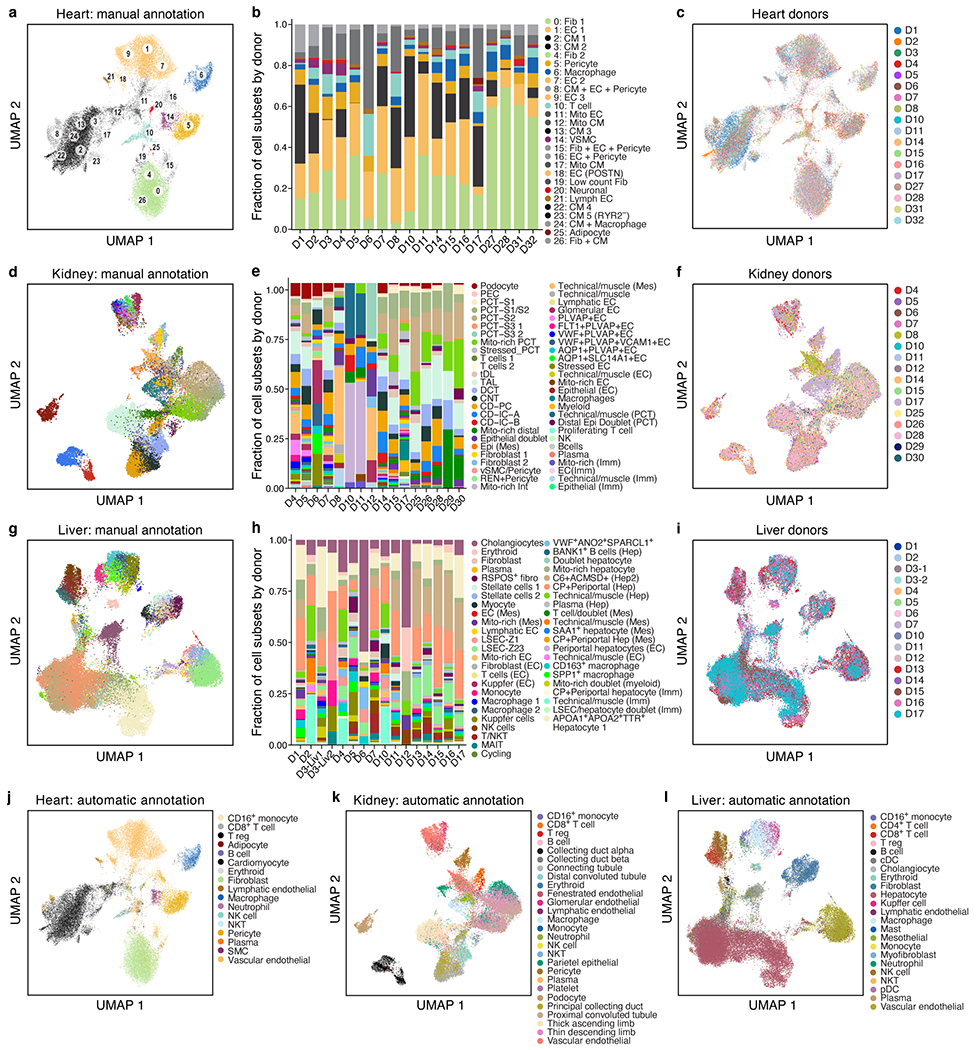
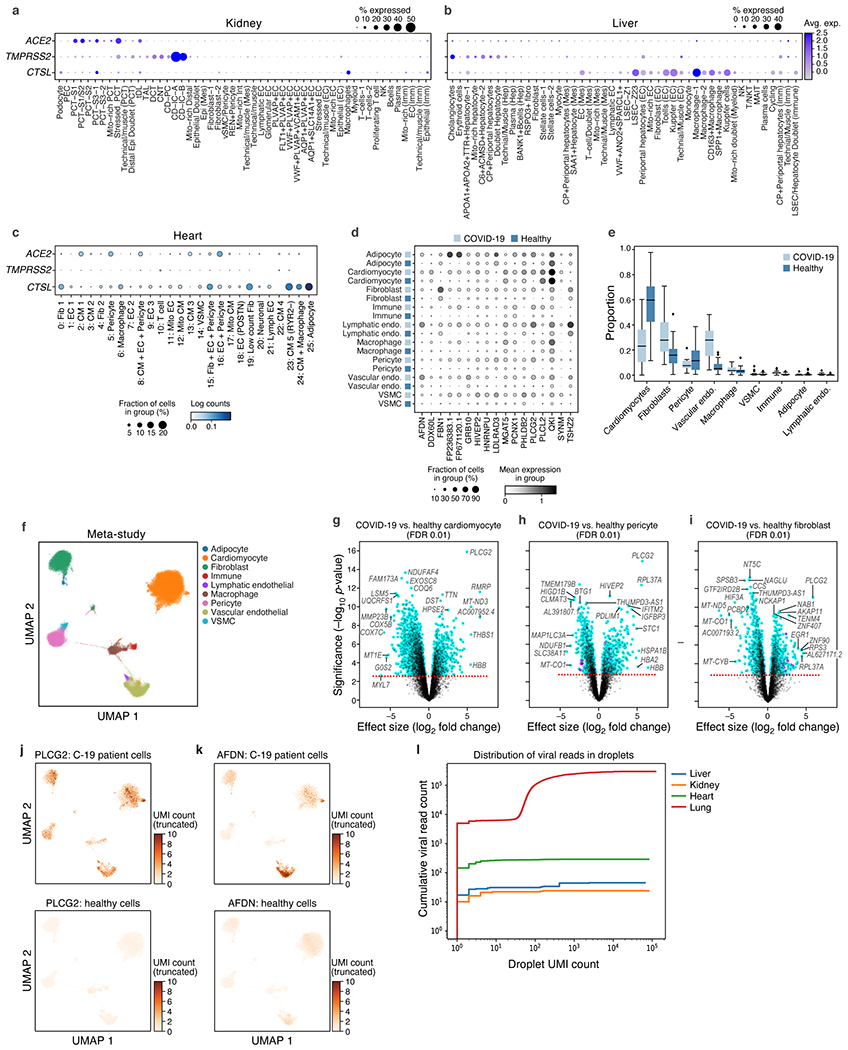
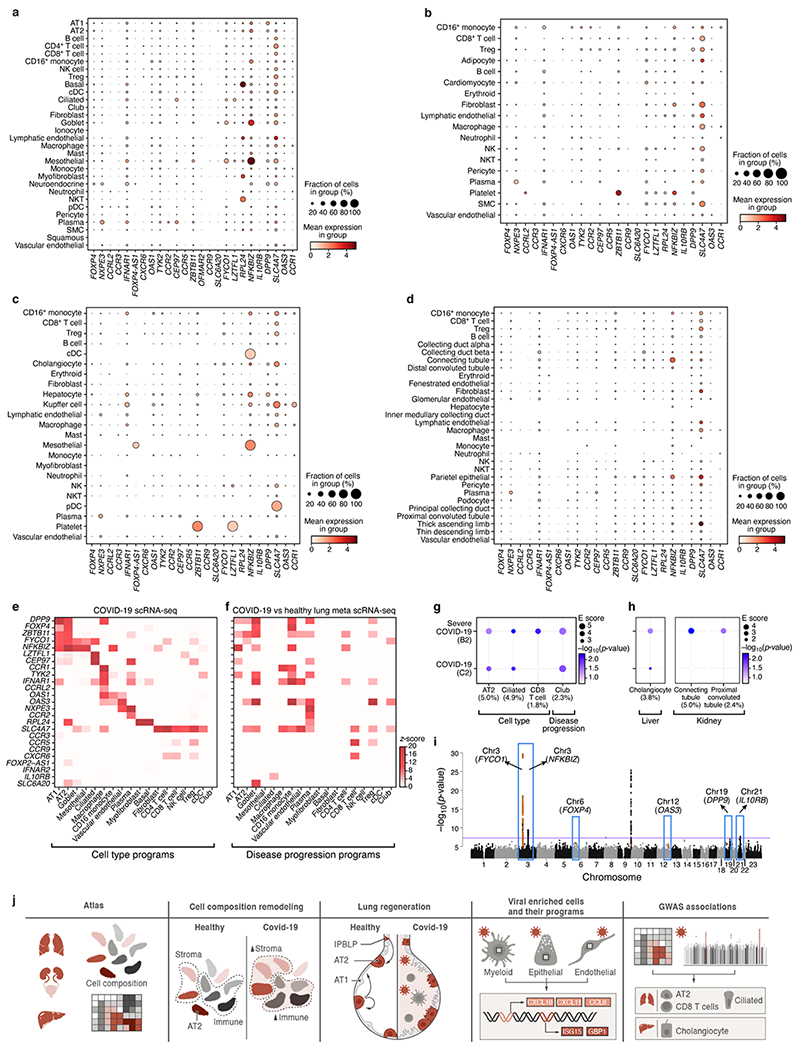
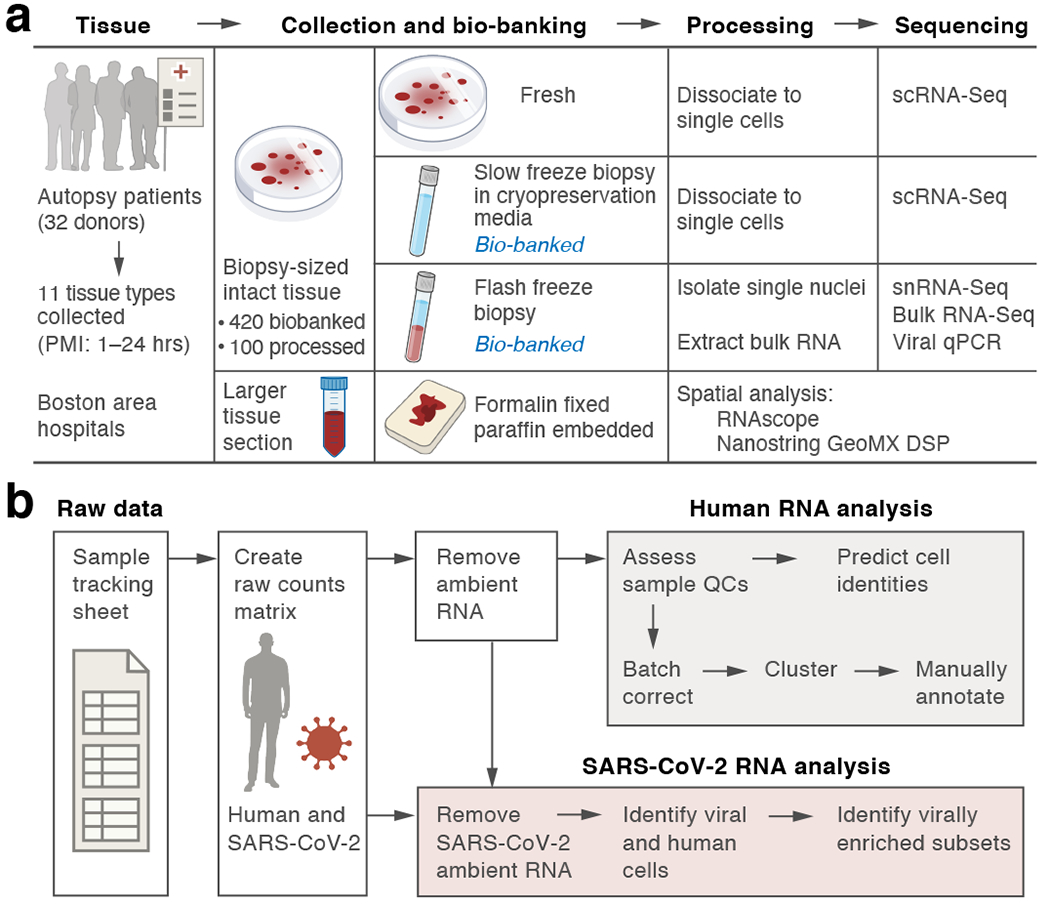
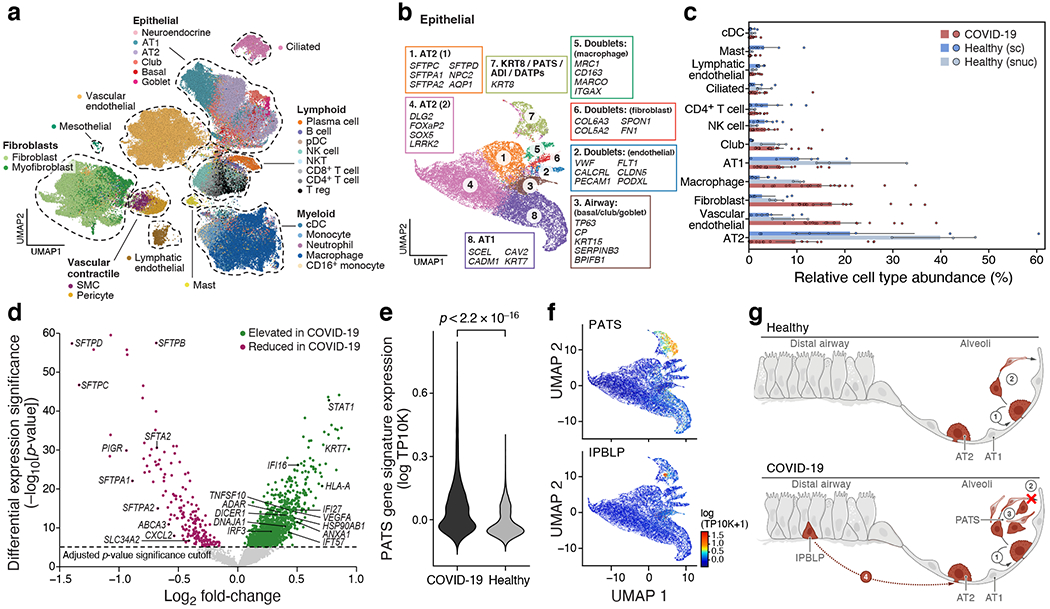
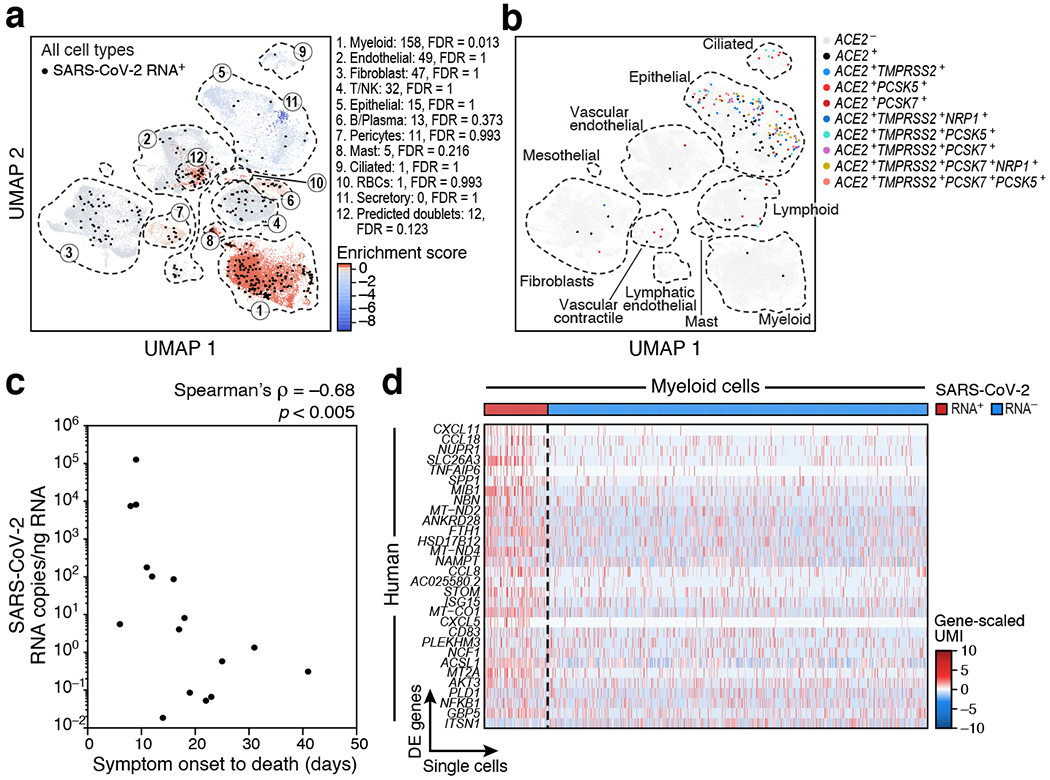
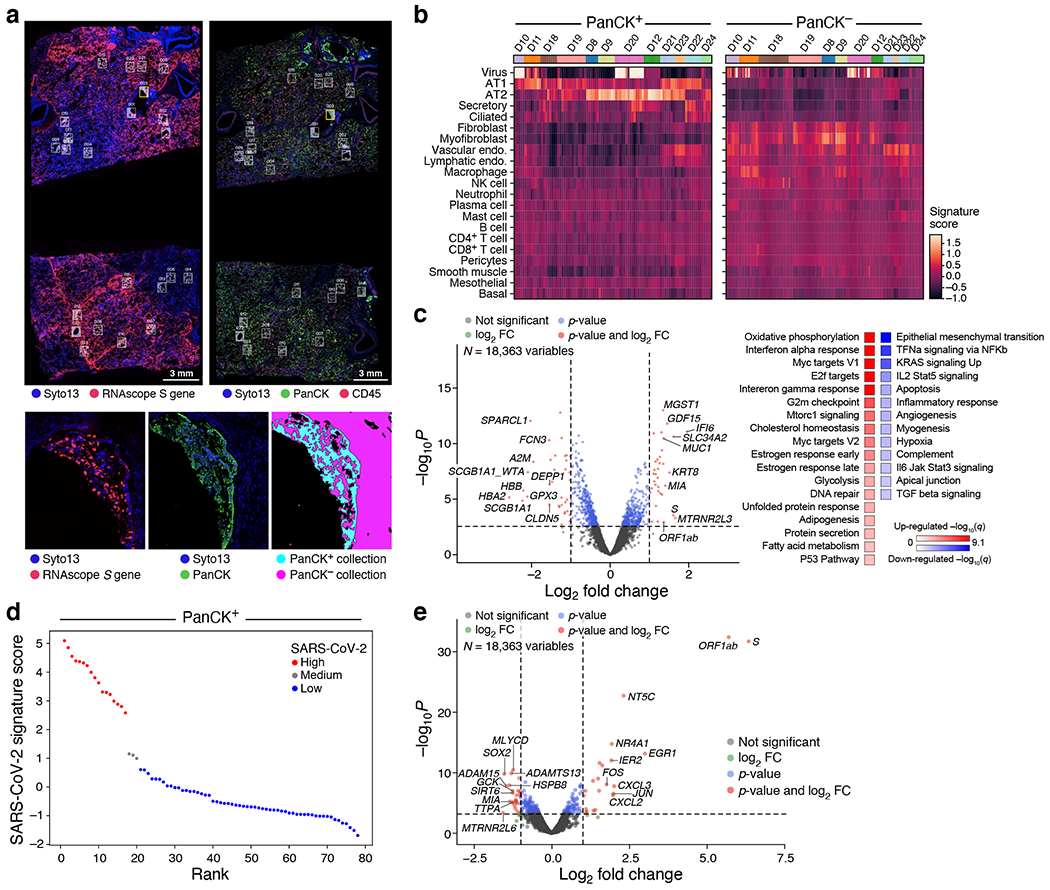
COVID-19, which is caused by SARS-CoV-2, can result in acute respiratory distress syndrome and multiple organ failure1-4, but little is known about its pathophysiology. Here we generated single-cell atlases of 24 lung, 16 kidney, 16 liver and 19 heart autopsy tissue samples and spatial atlases of 14 lung samples from donors who died of COVID-19. Integrated computational analysis uncovered substantial remodelling in the lung epithelial, immune and stromal compartments, with evidence of multiple paths of failed tissue regeneration, including defective alveolar type 2 differentiation and expansion of fibroblasts and putative TP63+ intrapulmonary basal-like progenitor cells. Viral RNAs were enriched in mononuclear phagocytic and endothelial lung cells, which induced specific host programs. Spatial analysis in lung distinguished inflammatory host responses in lung regions with and without viral RNA. Analysis of the other tissue atlases showed transcriptional alterations in multiple cell types in heart tissue from donors with COVID-19, and mapped cell types and genes implicated with disease severity based on COVID-19 genome-wide association studies. Our foundational dataset elucidates the biological effect of severe SARS-CoV-2 infection across the body, a key step towards new treatments.
Figures
Comment in
-
Single-cell atlases dissect tissue destruction by SARS-CoV-2.Nat Rev Immunol. 2021 Jun;21(6):343. doi: 10.1038/s41577-021-00564-5. Nat Rev Immunol. 2021. PMID: 33990788 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- WT_/Wellcome Trust/United Kingdom
- R37 MH107649/MH/NIMH NIH HHS/United States
- K08 CA222663/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- P30 DK046200/DK/NIDDK NIH HHS/United States
- R01 AA017729/AA/NIAAA NIH HHS/United States
- P30 CA013696/CA/NCI NIH HHS/United States
- UH3 HL141797/HL/NHLBI NIH HHS/United States
- R01 MH107649/MH/NIMH NIH HHS/United States
- R01 MH101244/MH/NIMH NIH HHS/United States
- R01 AA020744/AA/NIAAA NIH HHS/United States
- R37 CA258829/CA/NCI NIH HHS/United States
- U54 CA225088/CA/NCI NIH HHS/United States
- U01 AA026933/AA/NIAAA NIH HHS/United States
- P30 DK043351/DK/NIDDK NIH HHS/United States
- U01 HG009379/HG/NHGRI NIH HHS/United States
- DP2 CA247831/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
