

Identification of a Resistance Mechanism to IGF-IR Targeting in Human Triple Negative MDA-MB-231 Breast Cancer Cells
- PMID: 33916323
- PMCID: PMC8065809
- DOI: 10.3390/biom11040527
Identification of a Resistance Mechanism to IGF-IR Targeting in Human Triple Negative MDA-MB-231 Breast Cancer Cells
Abstract
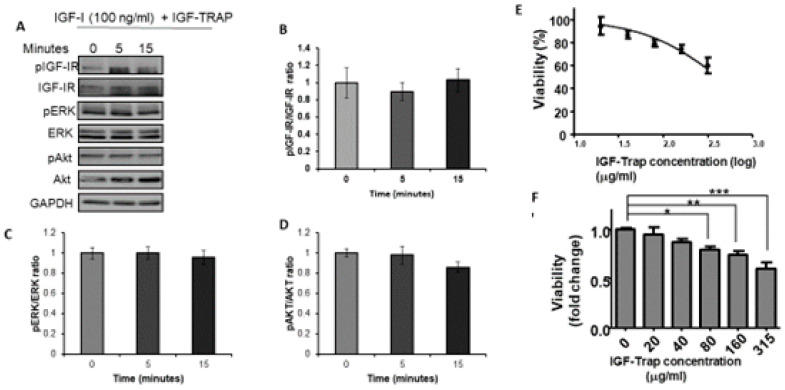
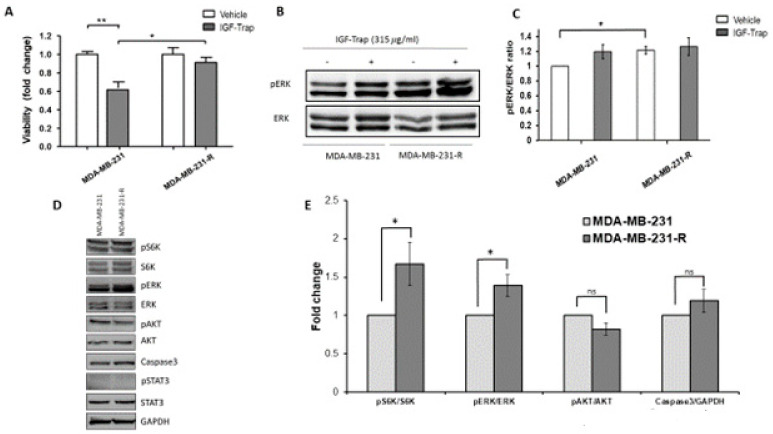
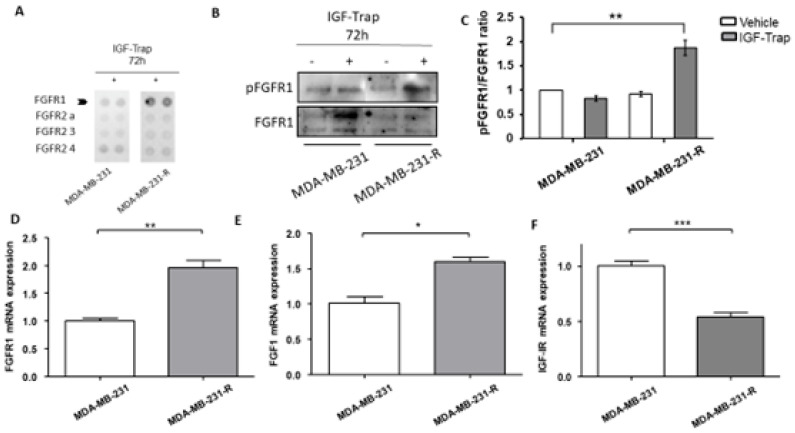
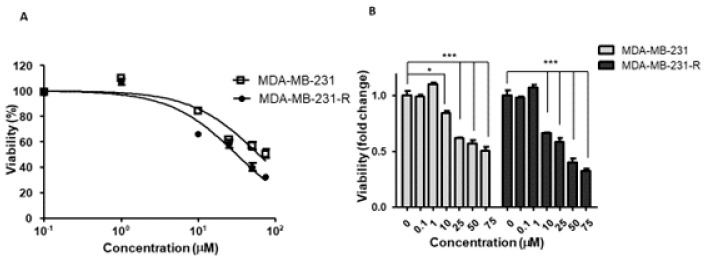
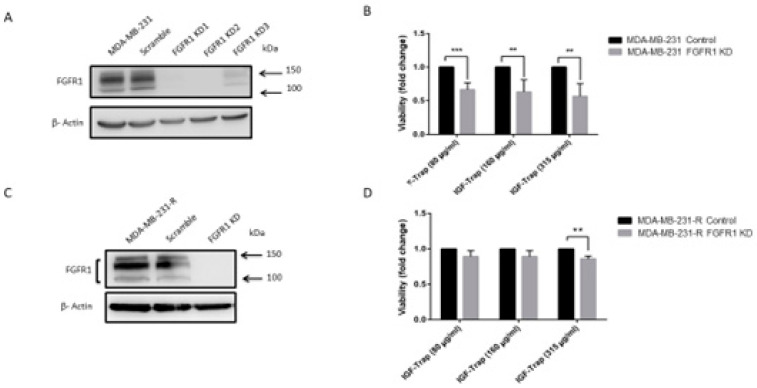
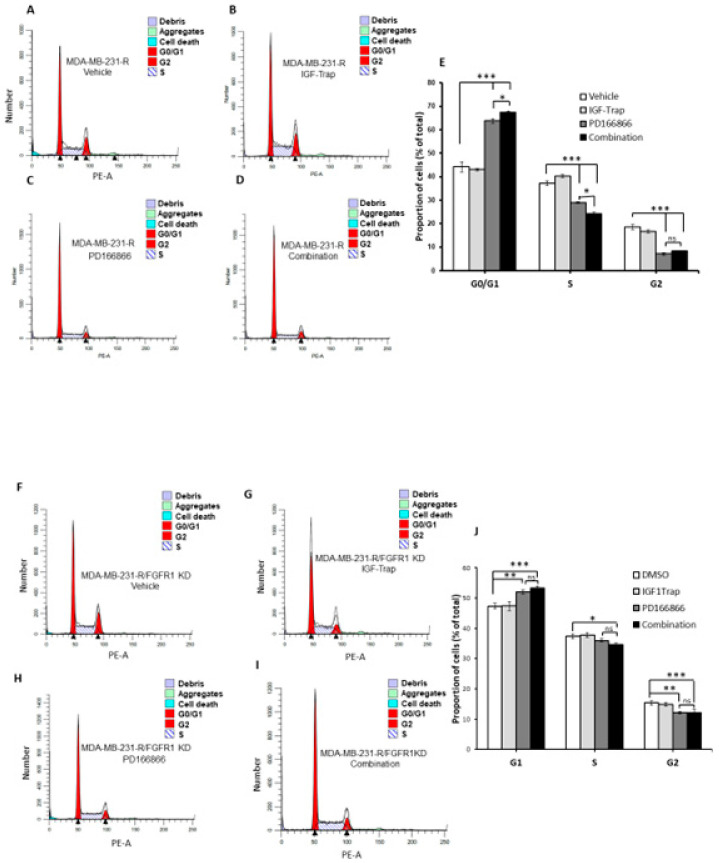
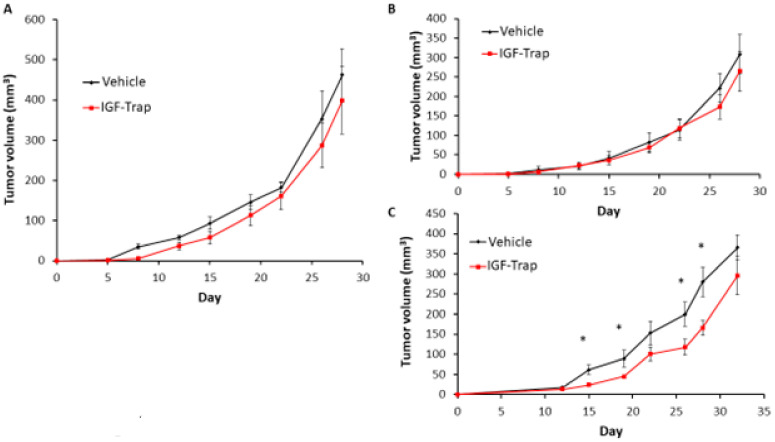
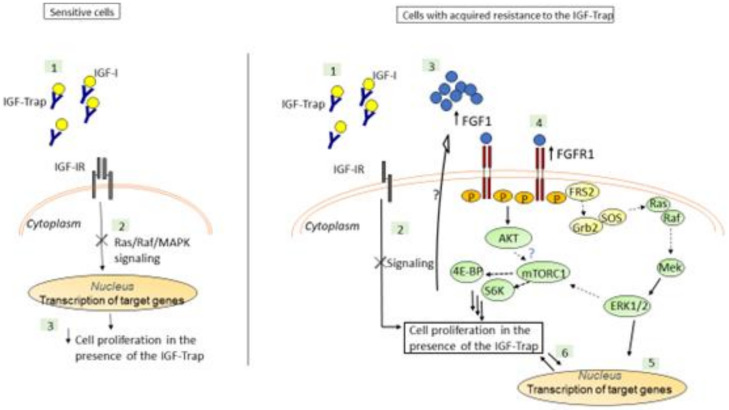
Triple negative breast cancer (TNBC) is associated with unfavorable prognosis and high relapse rates following chemotherapy. There is an urgent need to develop effective targeted therapy for this BC subtype. The type I insulin-like growth factor receptor (IGF-IR) was identified as a potential target for BC management. We previously reported on the production of the IGF-Trap, a soluble IGF-1R fusion protein that reduces the bioavailability of circulating IGF-1 and IGF-2 to the cognate receptor, impeding signaling. In nude mice xenotransplanted with the human TNBC MDA-MB-231 cells, we found variable responses to this inhibitor. We used this model to investigate potential resistance mechanisms to IGF-targeted therapy. We show here that prolonged exposure of MDA-MB-231 cells to the IGF-Trap in vitro selected a resistant subpopulation that proliferated unhindered in the presence of the IGF-Trap. We identified in these cells increased fibroblast growth factor receptor 1 (FGFR1) activation levels that sensitized them to the FGFR1-specific tyrosine kinase inhibitor PD166866. Treatment with this inhibitor caused cell cycle arrest in both the parental and resistant cells, markedly increasing cell death in the latter. When combined with the IGF-Trap, an increase in cell cycle arrest was observed in the resistant cells. Moreover, FGFR1 silencing increased the sensitivity of these cells to IGF-Trap treatment in vivo. Our data identify increased FGFR1 signaling as a resistance mechanism to targeted inhibition of the IGF-IR and suggest that dual IGF-1R/FGFR1 blockade may be required to overcome TNBC cell resistance to IGF-axis inhibitors.
Keywords: FGFR1; IGF signaling; drug resistance; the IGF-Trap; triple negative breast cancer.
Conflict of interest statement
The authors have no conflict of interest to report.
Figures



References
-
- Badve S., Dabbs D.J., Schnitt S.J., Baehner F.L., Decker T., Eusebi V., Fox S.B., Ichihara S., Jacquemier J., Lakhani S.R., et al. Basal-like and tri-ple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011;24:157–167. doi: 10.1038/modpathol.2010.200. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
