

Is the Antidepressant Activity of Selective Serotonin Reuptake Inhibitors Mediated by Nicotinic Acetylcholine Receptors?
- PMID: 33917953
- PMCID: PMC8068400
- DOI: 10.3390/molecules26082149
Is the Antidepressant Activity of Selective Serotonin Reuptake Inhibitors Mediated by Nicotinic Acetylcholine Receptors?
Abstract
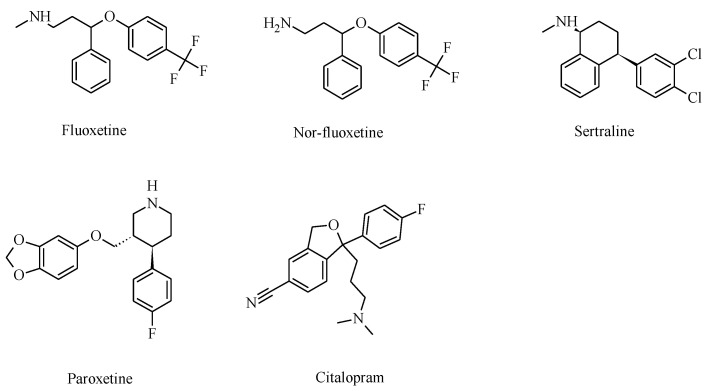
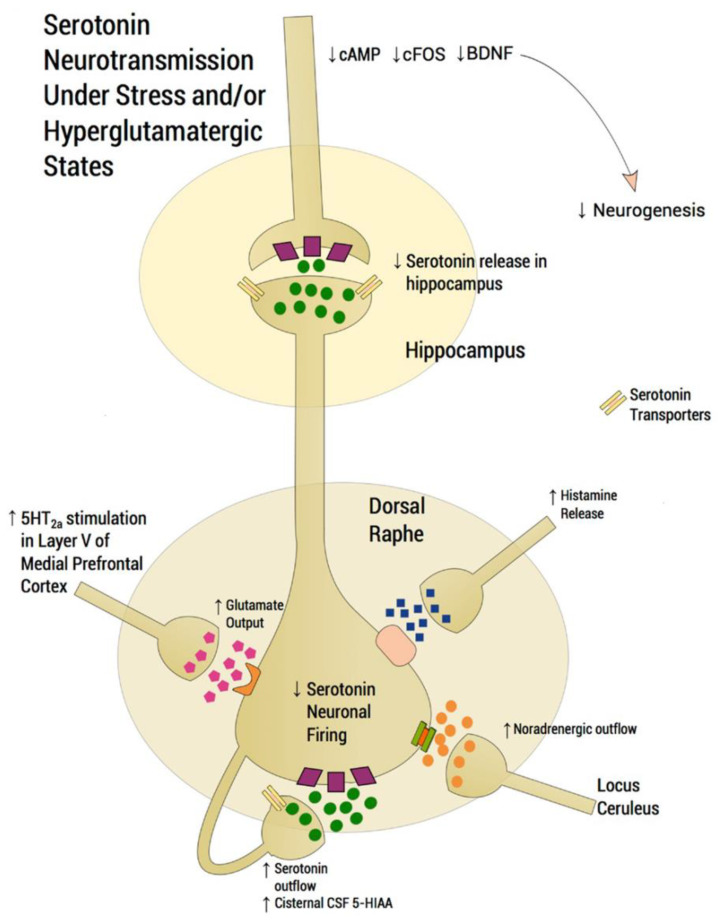
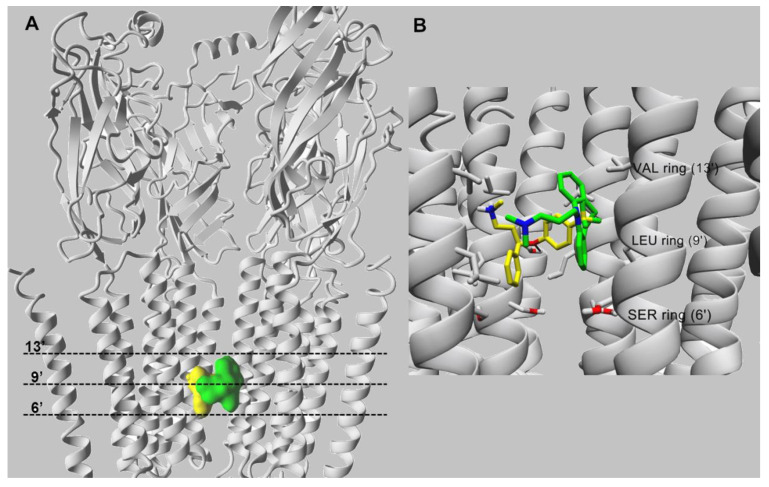
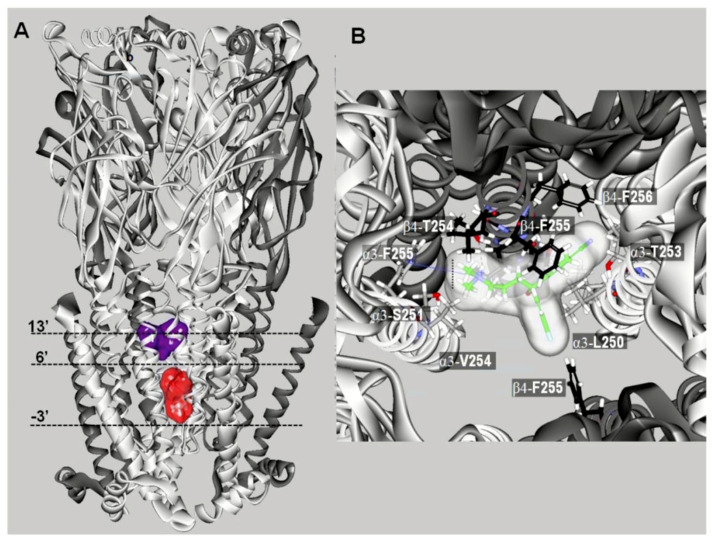
It is generally assumed that selective serotonin reuptake inhibitors (SSRIs) induce antidepressant activity by inhibiting serotonin (5-HT) reuptake transporters, thus elevating synaptic 5-HT levels and, finally, ameliorates depression symptoms. New evidence indicates that SSRIs may also modulate other neurotransmitter systems by inhibiting neuronal nicotinic acetylcholine receptors (nAChRs), which are recognized as important in mood regulation. There is a clear and strong association between major depression and smoking, where depressed patients smoke twice as much as the normal population. However, SSRIs are not efficient for smoking cessation therapy. In patients with major depressive disorder, there is a lower availability of functional nAChRs, although their amount is not altered, which is possibly caused by higher endogenous ACh levels, which consequently induce nAChR desensitization. Other neurotransmitter systems have also emerged as possible targets for SSRIs. Studies on dorsal raphe nucleus serotoninergic neurons support the concept that SSRI-induced nAChR inhibition decreases the glutamatergic hyperstimulation observed in stress conditions, which compensates the excessive 5-HT overflow in these neurons and, consequently, ameliorates depression symptoms. At the molecular level, SSRIs inhibit different nAChR subtypes by noncompetitive mechanisms, including ion channel blockade and induction of receptor desensitization, whereas α9α10 nAChRs, which are peripherally expressed and not directly involved in depression, are inhibited by competitive mechanisms. According to the functional and structural results, SSRIs bind within the nAChR ion channel at high-affinity sites that are spread out between serine and valine rings. In conclusion, SSRI-induced inhibition of a variety of nAChRs expressed in different neurotransmitter systems widens the complexity by which these antidepressants may act clinically.
Keywords: antidepressants; molecular modeling; neuronal pathways; nicotinic acetylcholine receptors; noncompetitive antagonists; selective serotonin reuptake inhibitors.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
References
-
- DSM-IV . Diagnostic and Statistical Manual of Mental Disorders IV-TR. American Psychiatric Press; Washington, DC, USA: 2020.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
