

MCAT Mutations Cause Nuclear LHON-like Optic Neuropathy
- PMID: 33918393
- PMCID: PMC8067165
- DOI: 10.3390/genes12040521
MCAT Mutations Cause Nuclear LHON-like Optic Neuropathy
Abstract
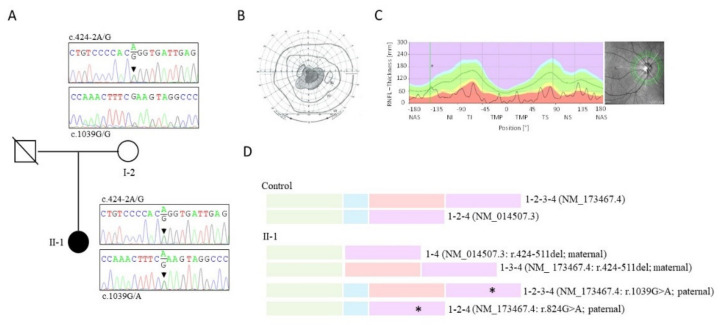
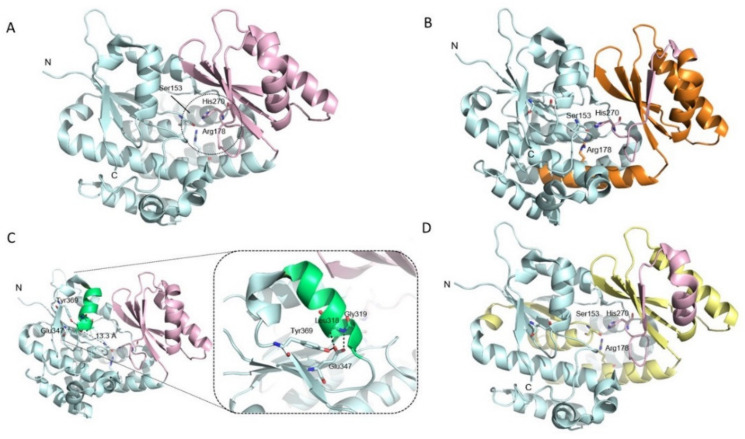
Pathological variants in the nuclear malonyl-CoA-acyl carrier protein transacylase (MCAT) gene, which encodes a mitochondrial protein involved in fatty-acid biogenesis, have been reported in two siblings from China affected by insidious optic nerve degeneration in childhood, leading to blindness in the first decade of life. After analysing 51 families with negative molecular diagnostic tests, from a cohort of 200 families with hereditary optic neuropathy (HON), we identified two novel MCAT mutations in a female patient who presented with acute, sudden, bilateral, yet asymmetric, central visual loss at the age of 20. This presentation is consistent with a Leber hereditary optic neuropathy (LHON)-like phenotype, whose existence and association with NDUFS2 and DNAJC30 has only recently been described. Our findings reveal a wider phenotypic presentation of MCAT mutations, and a greater genetic heterogeneity of nuclear LHON-like phenotypes. Although MCAT pathological variants are very uncommon, this gene should be investigated in HON patients, irrespective of disease presentation.
Keywords: MCAT; hereditary optic neuropathy (HON); nuclear LHON-like.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Yu-Wai-Man P., Chinnery P.F. In: Leber Hereditary Optic Neuropathy. Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Mirzaa G., Amemiya A., editors. University of Washington; Seattle, WA, USA: 1993–2021. GeneReviews® [Internet]
-
- Delettre C., Lenaers G., Griffoin J.-M., Gigarel N., Lorenzo C., Belenguer P., Pelloquin L., Grosgeorge J., Turc-Carel C., Perret E., et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000;26:207–210. doi: 10.1038/79936. - DOI - PubMed
-
- Alexander C., Votruba M., Pesch U.E., Thiselton D.L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G., et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000;26:211–215. doi: 10.1038/79944. - DOI - PubMed
-
- Gerber S., Charif M., Chevrollier A., Chaumette T., Angebault C., Kane M.S., Paris A., Alban J., Quiles M., Delettre C., et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain. 2017;140:2586–2596. doi: 10.1093/brain/awx219. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
