

Integrated Metabolomics and Transcriptomics Using an Optimised Dual Extraction Process to Study Human Brain Cancer Cells and Tissues
- PMID: 33919944
- PMCID: PMC8070957
- DOI: 10.3390/metabo11040240
Integrated Metabolomics and Transcriptomics Using an Optimised Dual Extraction Process to Study Human Brain Cancer Cells and Tissues
Abstract
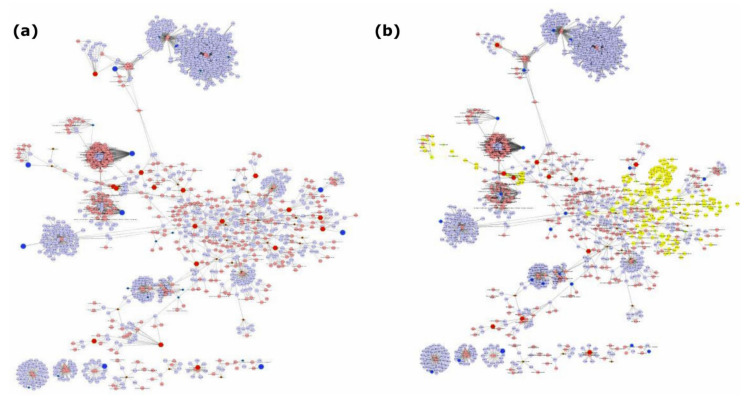
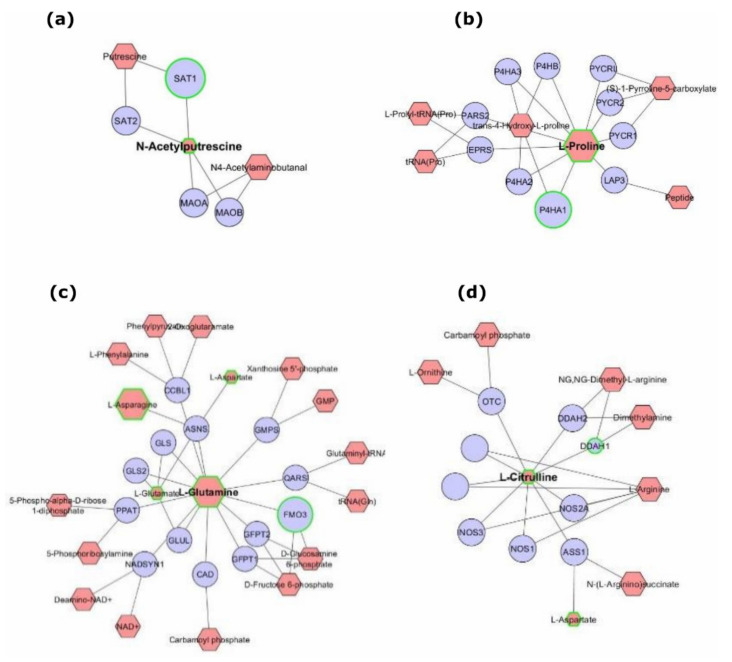
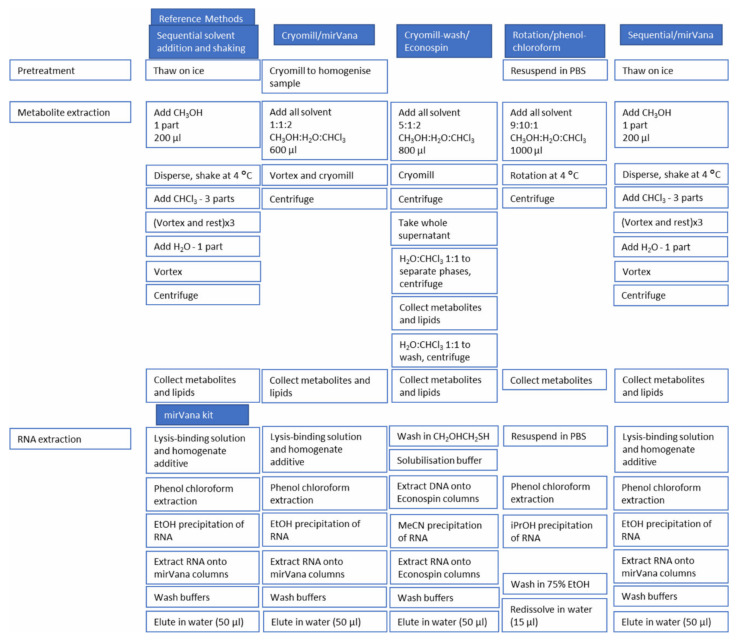
The integration of untargeted metabolomics and transcriptomics from the same population of cells or tissue enhances the confidence in the identified metabolic pathways and understanding of the enzyme-metabolite relationship. Here, we optimised a simultaneous extraction method of metabolites/lipids and RNA from ependymoma cells (BXD-1425). Relative to established RNA (mirVana kit) or metabolite (sequential solvent addition and shaking) single extraction methods, four dual-extraction techniques were evaluated and compared (methanol:water:chloroform ratios): cryomill/mirVana (1:1:2); cryomill-wash/Econospin (5:1:2); rotation/phenol-chloroform (9:10:1); Sequential/mirVana (1:1:3). All methods extracted the same metabolites, yet rotation/phenol-chloroform did not extract lipids. Cryomill/mirVana and sequential/mirVana recovered the highest amounts of RNA, at 70 and 68% of that recovered with mirVana kit alone. sequential/mirVana, involving RNA extraction from the interphase of our established sequential solvent addition and shaking metabolomics-lipidomics extraction method, was the most efficient approach overall. Sequential/mirVana was applied to study a) the biological effect caused by acute serum starvation in BXD-1425 cells and b) primary ependymoma tumour tissue. We found (a) 64 differentially abundant metabolites and 28 differentially expressed metabolic genes, discovering four gene-metabolite interactions, and (b) all metabolites and 62% lipids were above the limit of detection, and RNA yield was sufficient for transcriptomics, in just 10 mg of tissue.
Keywords: RNA; cancer; dual-extraction; integrated omics; metabolite.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
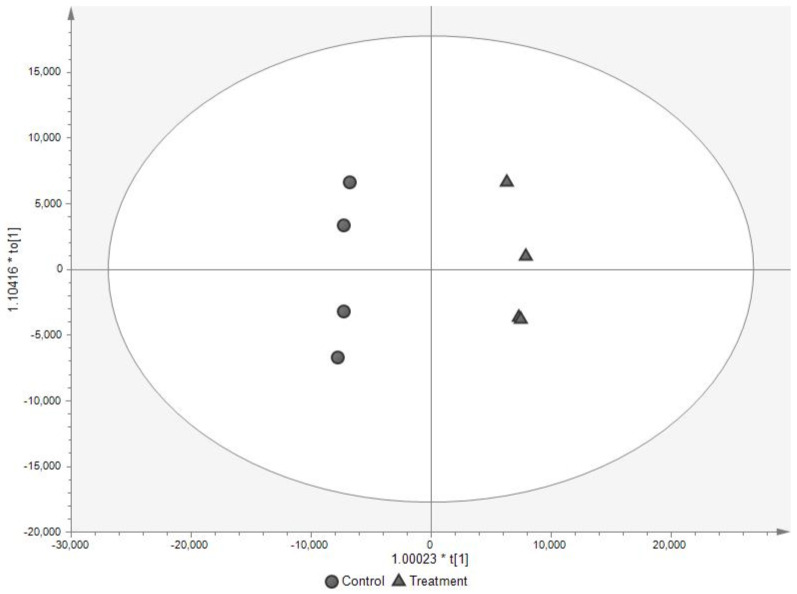
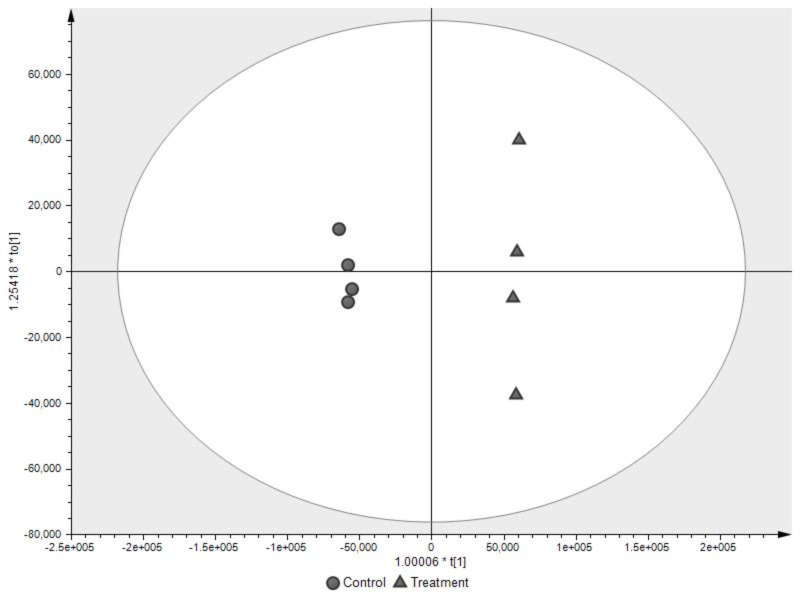
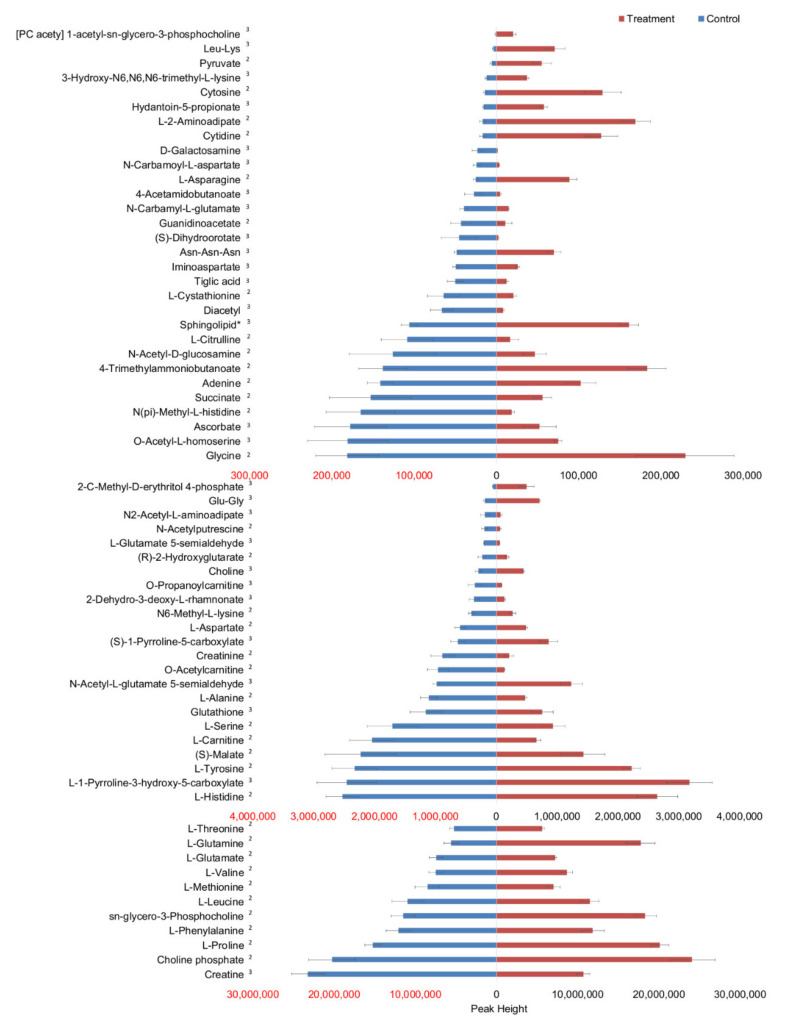
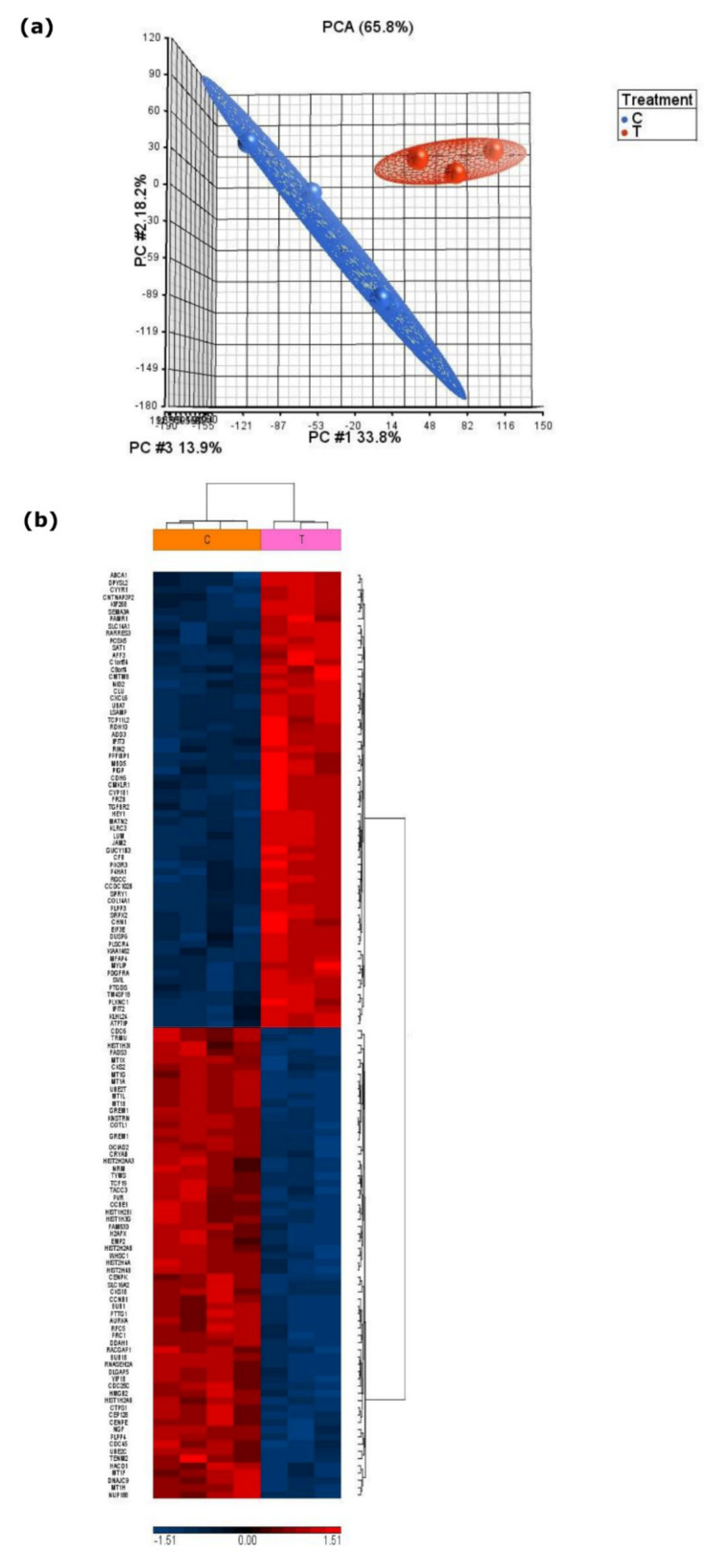
References
-
- Karnovsky A., Weymouth T., Hull T., Glenn Tarcea V., Scardoni G., Laudanna C., Sartor M.A., Stringer K.A., Jagadish H.V., Burant C., et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics. 2012;28:373–380. doi: 10.1093/bioinformatics/btr661. - DOI - PMC - PubMed
-
- Futschik M.E., Morkel M., Schäfer R., Sers C. In: The Human Transcriptome: Implications for Understanding, Diagnosing, and Treating Human Disease. 2nd ed. Coleman W.B., Tsongalis G.J., editors. Academic Press; Cambridge, MA, USA: 2018. pp. 135–164.
-
- Challacombe J.F. In silico identification of metabolic enzyme drug targets in Burkholderia pseudomallei. bioRxiv. 2017:1–33. doi: 10.1101/034306. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
