

Combining Functional Genomics and Whole-Genome Sequencing to Detect Antibiotic Resistance Genes in Bacterial Strains Co-Occurring Simultaneously in a Brazilian Hospital
- PMID: 33920372
- PMCID: PMC8070361
- DOI: 10.3390/antibiotics10040419
Combining Functional Genomics and Whole-Genome Sequencing to Detect Antibiotic Resistance Genes in Bacterial Strains Co-Occurring Simultaneously in a Brazilian Hospital
Abstract
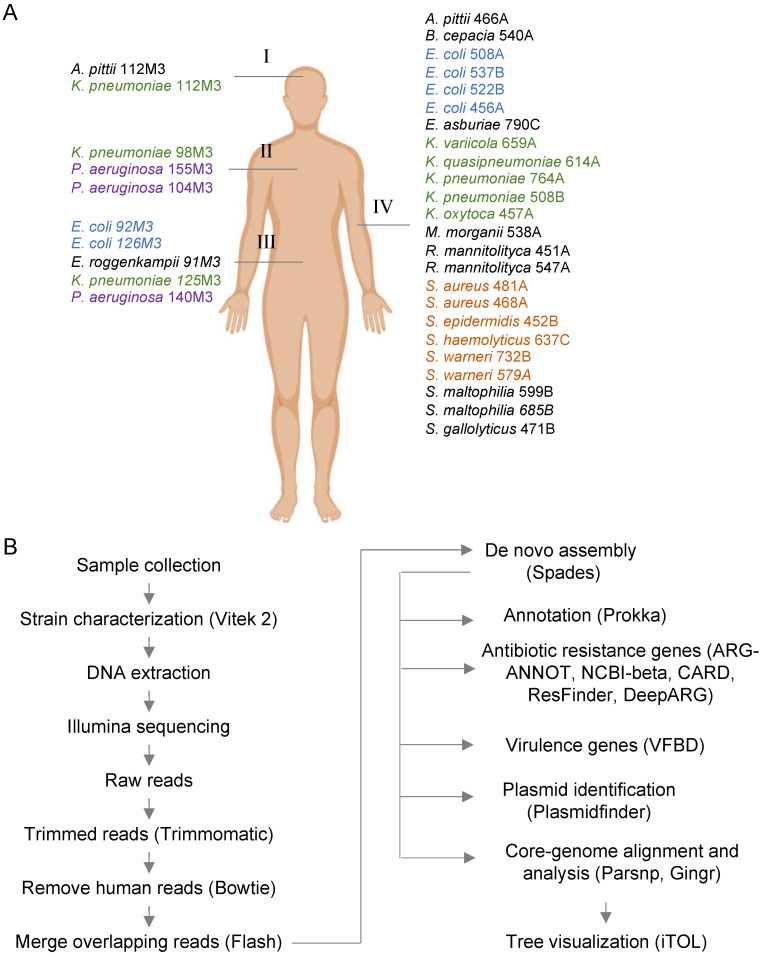
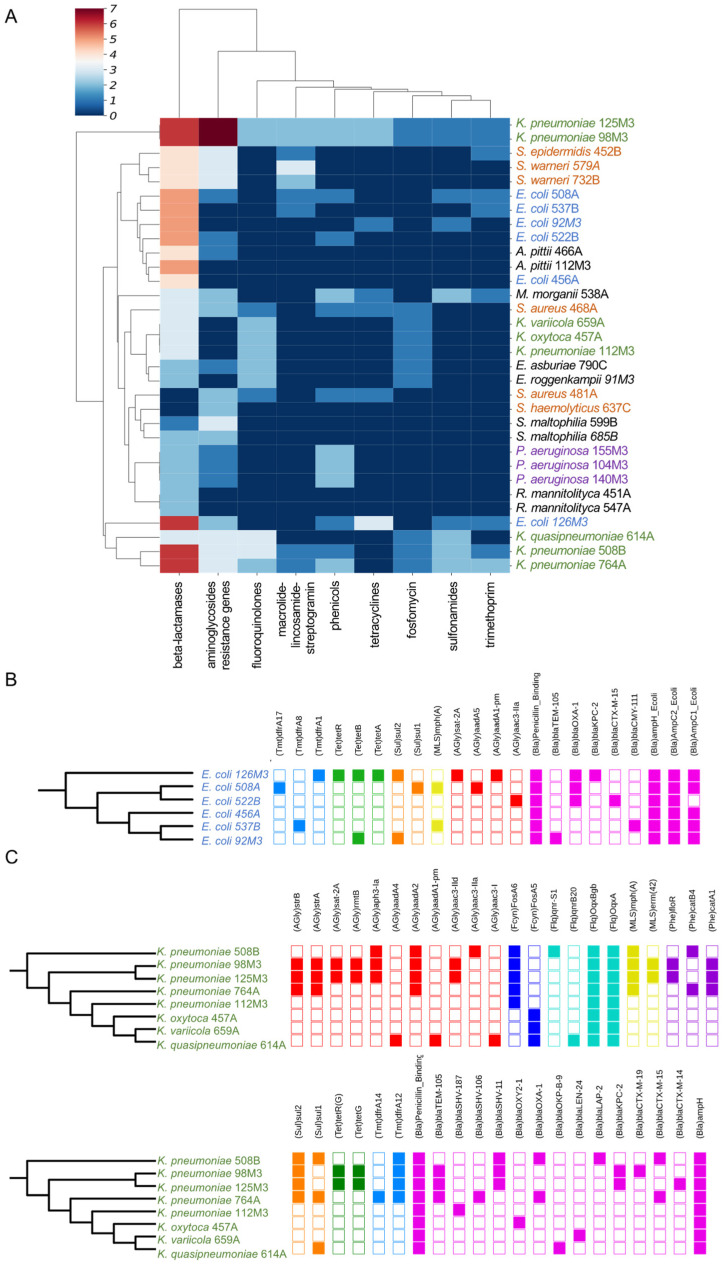
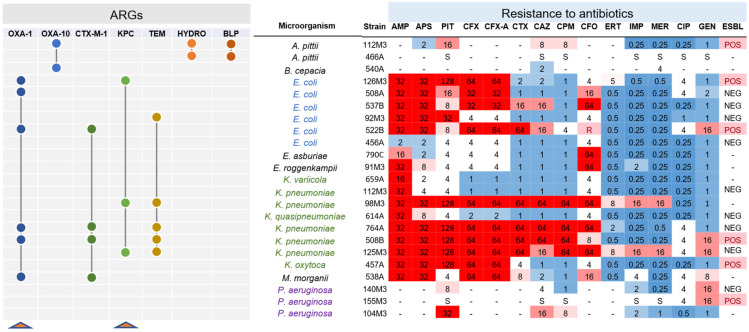
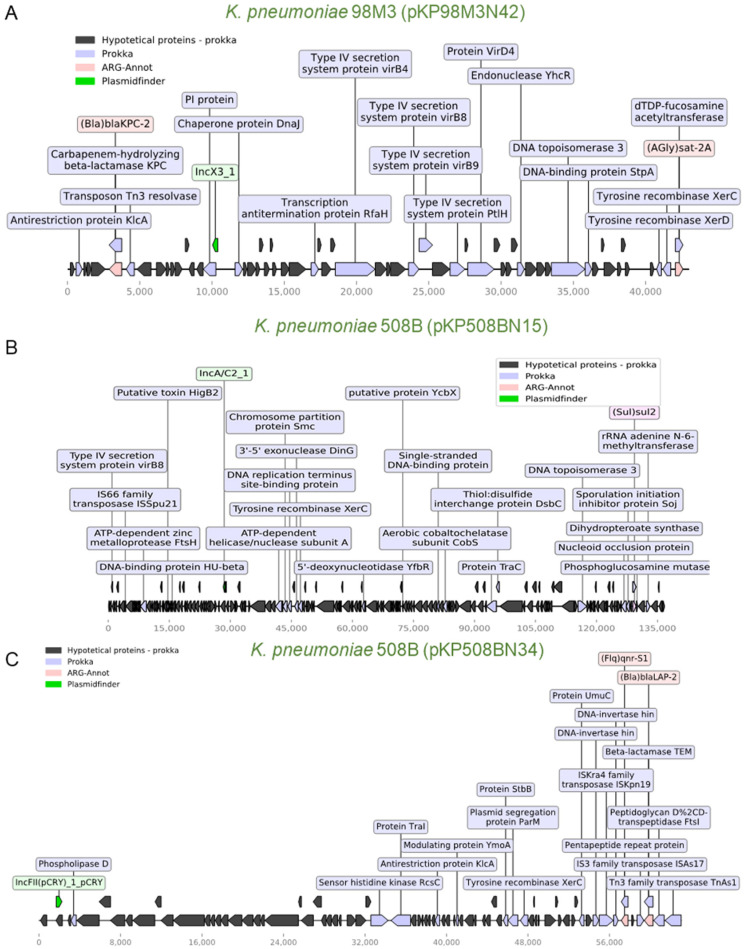
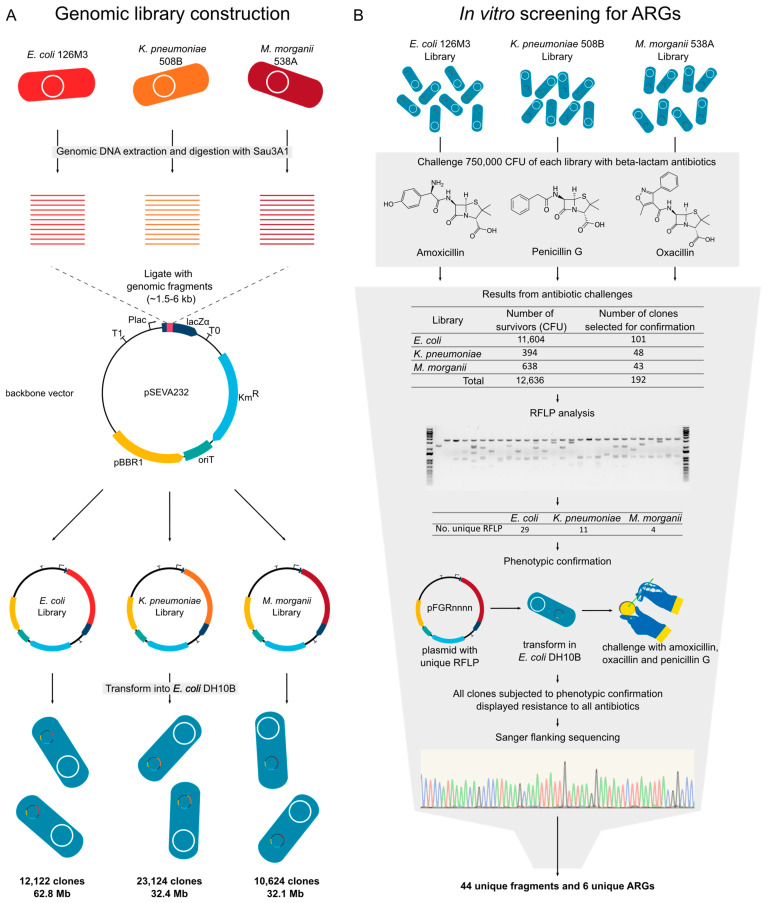
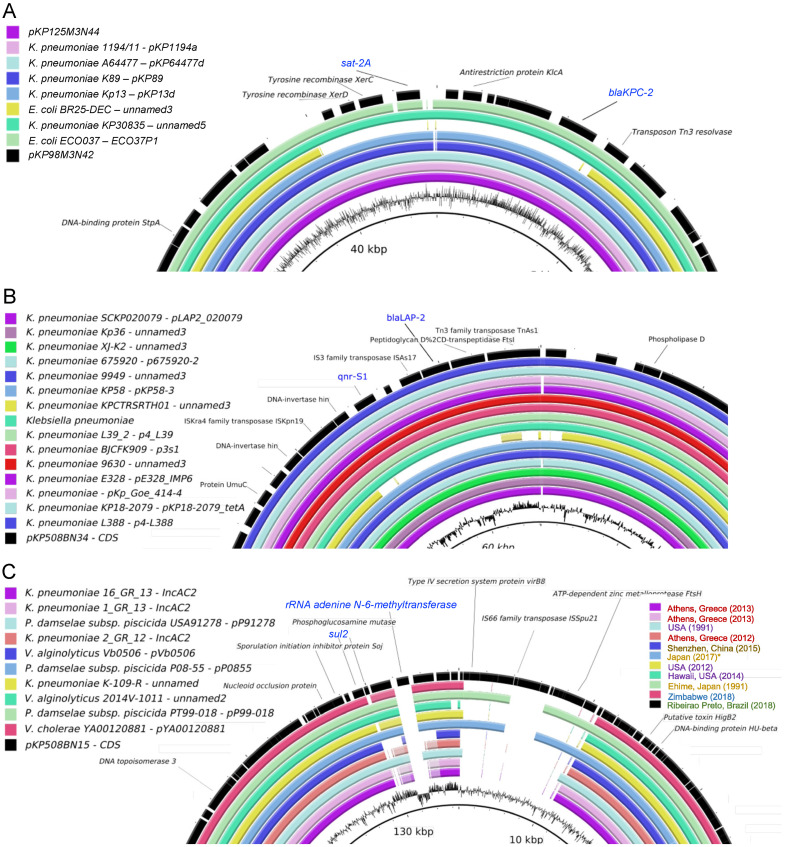
(1) Background: The rise of multi-antibiotic resistant bacteria represents an emergent threat to human health. Here, we investigate antibiotic resistance mechanisms in bacteria of several species isolated from an intensive care unit in Brazil. (2) Methods: We used whole-genome analysis to identify antibiotic resistance genes (ARGs) and plasmids in 34 strains of Gram-negative and Gram-positive bacteria, providing the first genomic description of Morganella morganii and Ralstonia mannitolilytica clinical isolates from South America. (3) Results: We identified a high abundance of beta-lactamase genes in resistant organisms, including seven extended-spectrum beta-lactamases (OXA-1, OXA-10, CTX-M-1, KPC, TEM, HYDRO, BLP) shared between organisms from different species. Additionally, we identified several ARG-carrying plasmids indicating the potential for a fast transmission of resistance mechanism between bacterial strains. Furthermore, we uncovered two pairs of (near) identical plasmids exhibiting multi-drug resistance. Finally, since many highly resistant strains carry several different ARGs, we used functional genomics to investigate which of them were indeed functional. In this sense, for three bacterial strains (Escherichia coli, Klebsiella pneumoniae, and M. morganii), we identified six beta-lactamase genes out of 15 predicted in silico as those mainly responsible for the resistance mechanisms observed, corroborating the existence of redundant resistance mechanisms in these organisms. (4) Conclusions: Systematic studies similar to the one presented here should help to prevent outbreaks of novel multidrug-resistant bacteria in healthcare facilities.
Keywords: antibiotic resistance genes; functional genomics; mobilome; resistome; whole-genome analysis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Pancholi P., Carroll K.C., Buchan B.W., Chan R.C., Dhiman N., Ford B., Granato P.A., Harrington A.T., Hernandez D.R., Humphries R.M., et al. Multicenter Evaluation of the Accelerate PhenoTest BC Kit for Rapid Identification and Phenotypic Antimicrobial Susceptibility Testing Using Morphokinetic Cellular Analysis. J. Clin. Microbiol. 2018;56 doi: 10.1128/JCM.01329-17. - DOI - PMC - PubMed
-
- Köser C.U., Ellington M.J., Cartwright E.J.P., Gillespie S.H., Brown N.M., Farrington M., Holden M.T.G., Dougan G., Bentley S.D., Parkhill J., et al. Routine Use of Microbial Whole Genome Sequencing in Diagnostic and Public Health Microbiology. PLoS Pathog. 2012;8:e1002824. doi: 10.1371/journal.ppat.1002824. - DOI - PMC - PubMed
-
- Adams M.D., Goglin K., Molyneaux N., Hujer K.M., Lavender H., Jamison J.J., Macdonald I.J., Martin K.M., Russo T., Campagnari A.A., et al. Comparative Genome Sequence Analysis of Multidrug-Resistant Acinetobacter baumannii. J. Bacteriol. 2008;190:8053–8064. doi: 10.1128/JB.00834-08. - DOI - PMC - PubMed
Grants and funding
- 2015/04309-1/Fundação de Amparo à Pesquisa do Estado de São Paulo
- 2019/15675-0/Fundação de Amparo à Pesquisa do Estado de São Paulo
- 2019/00390-0/Fundação de Amparo à Pesquisa do Estado de São Paulo
- 2018/18158-3/Fundação de Amparo à Pesquisa do Estado de São Paulo
- 2016/18827-7/Fundação de Amparo à Pesquisa do Estado de São Paulo
LinkOut - more resources
Full Text Sources
Other Literature Sources
