

The Major Constituent of Green Tea, Epigallocatechin-3-Gallate (EGCG), Inhibits the Growth of HPV18-Infected Keratinocytes by Stimulating Proteasomal Turnover of the E6 and E7 Oncoproteins
- PMID: 33920477
- PMCID: PMC8069595
- DOI: 10.3390/pathogens10040459
The Major Constituent of Green Tea, Epigallocatechin-3-Gallate (EGCG), Inhibits the Growth of HPV18-Infected Keratinocytes by Stimulating Proteasomal Turnover of the E6 and E7 Oncoproteins
Abstract
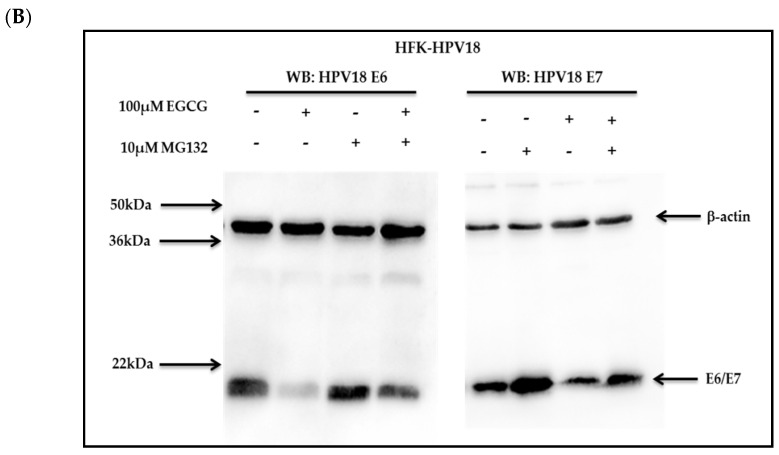
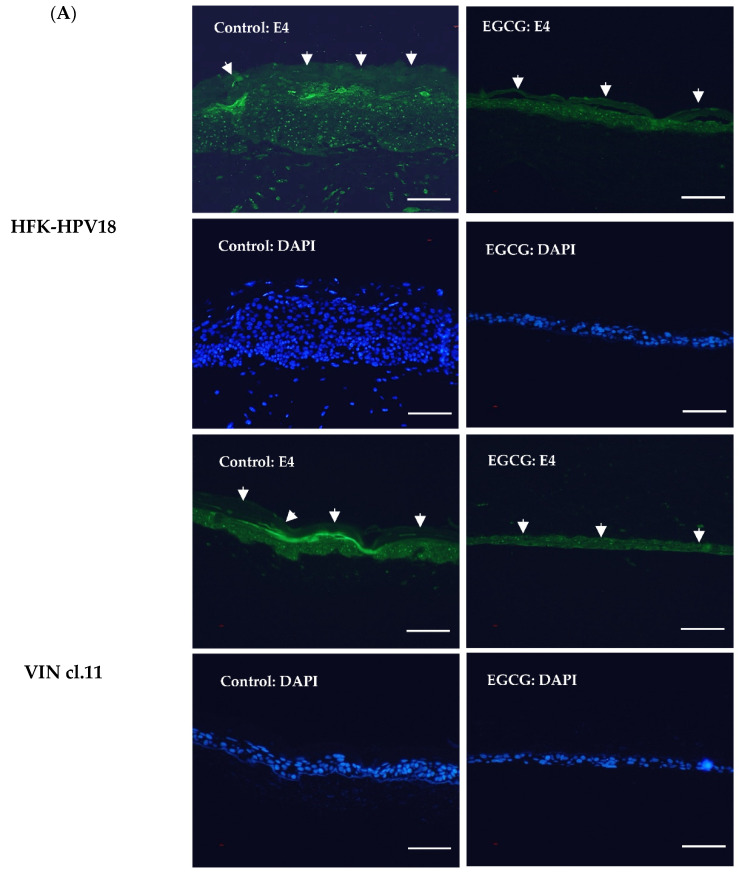
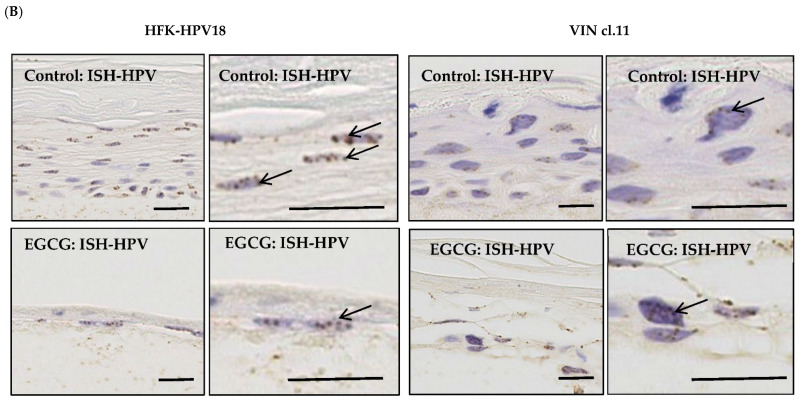
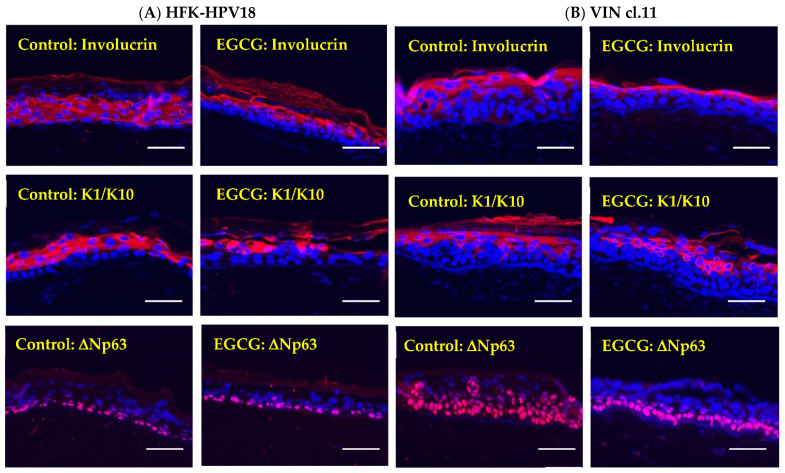
Epigallocatechin-3-gallate (EGCG), the primary bioactive polyphenol in green tea, has been shown to inhibit the growth of human papilloma virus (HPV)-transformed keratinocytes. Here, we set out to examine the consequences of EGCG treatment on the growth of HPV18-immortalised foreskin keratinocytes (HFK-HPV18) and an authentic HPV18-positive vulvar intraepithelial neoplasia (VIN) clone, focusing on its ability to influence cell proliferation and differentiation and to impact on viral oncogene expression and virus replication. EGCG treatment was associated with degradation of the E6 and E7 oncoproteins and an upregulation of their associated tumour suppressor genes; consequently, keratinocyte proliferation was inhibited in both monolayer and organotypic raft culture. While EGCG exerted a profound effect on cell proliferation, it had little impact on keratinocyte differentiation. Expression of the late viral protein E4 was suppressed in the presence of EGCG, suggesting that EGCG was able to block productive viral replication in differentiating keratinocytes. Although EGCG did not alter the levels of E6 and E7 mRNA, it enhanced the turnover of the E6 and E7 proteins. The addition of MG132, a proteasome inhibitor, to EGCG-treated keratinocytes led to the accumulation of the E6/E7 proteins, showing that EGCG acts as an anti-viral, targeting the E6 and E7 proteins for proteasome-mediated degradation.
Keywords: epigallocatechin-3-gallate; human papilloma virus; vulval intraepithelial neoplasia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
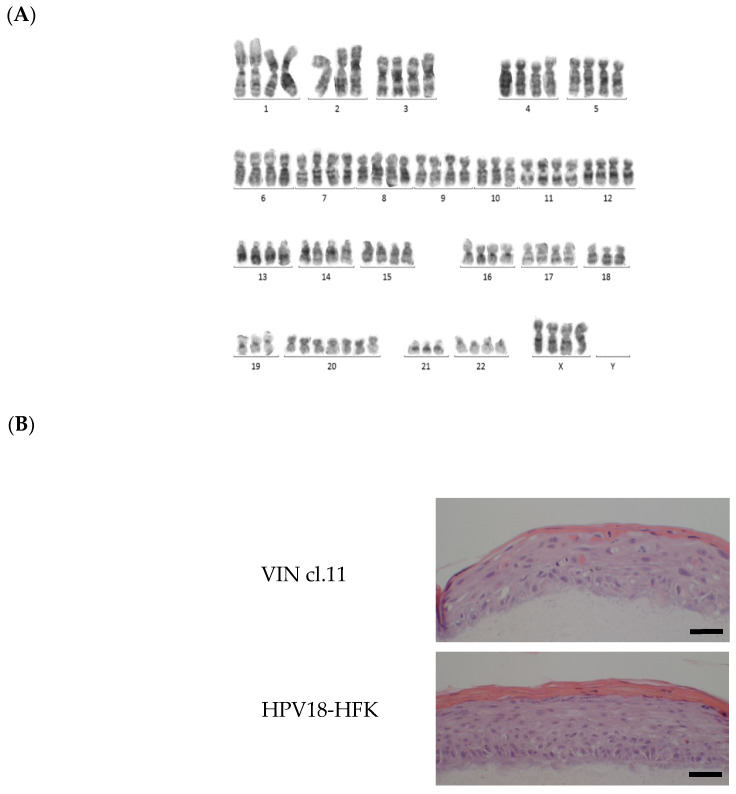
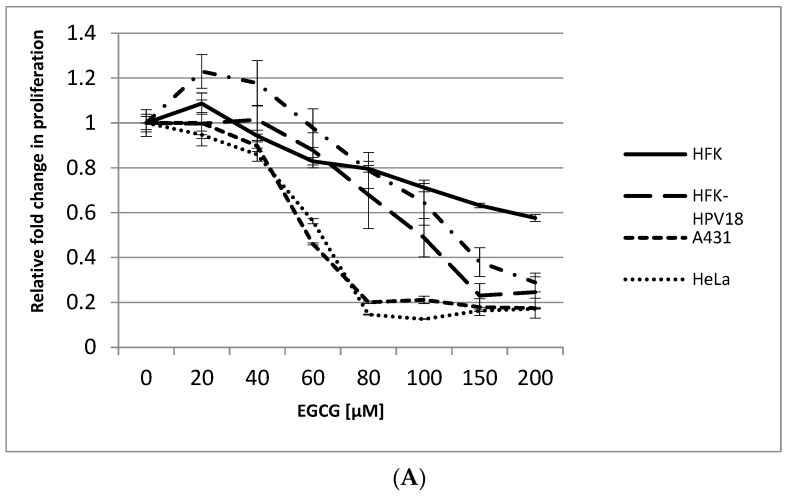
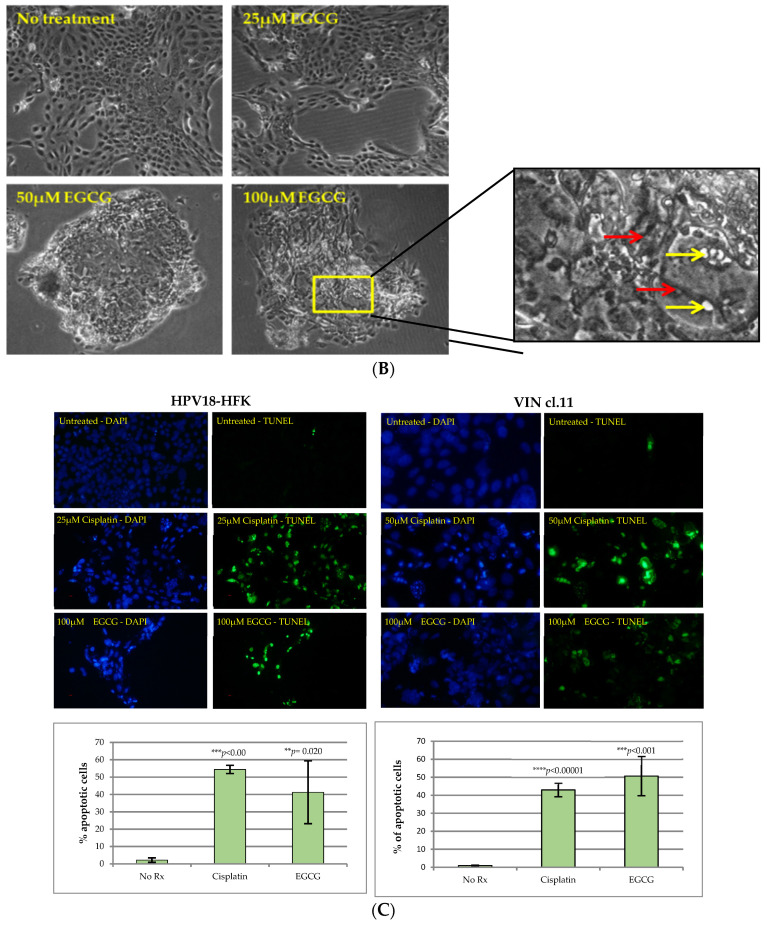
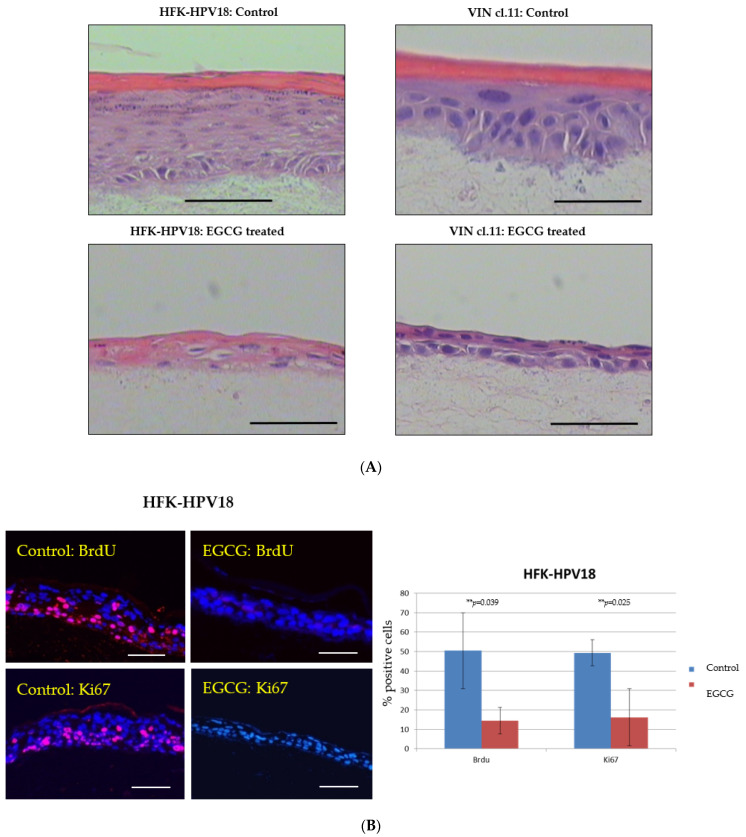
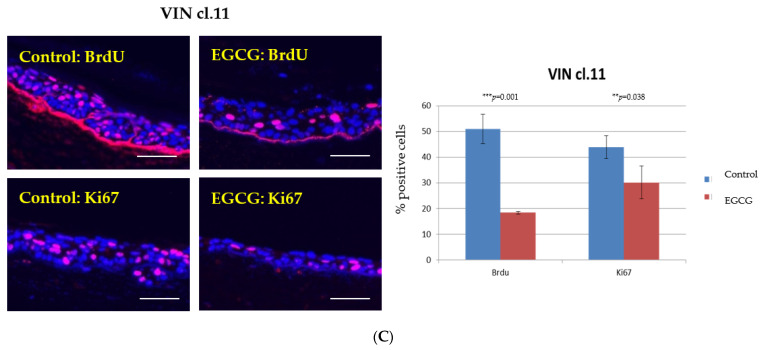
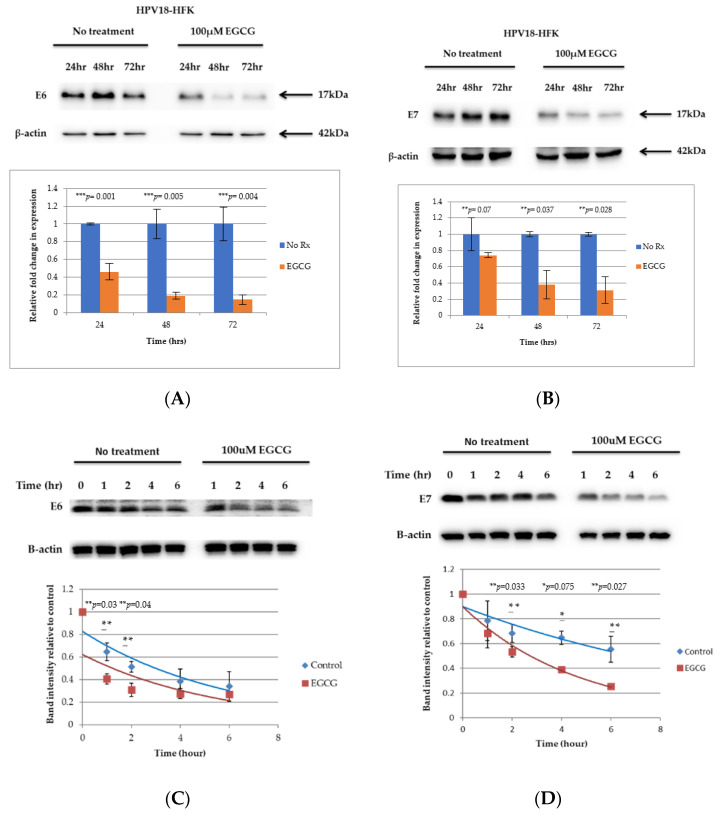
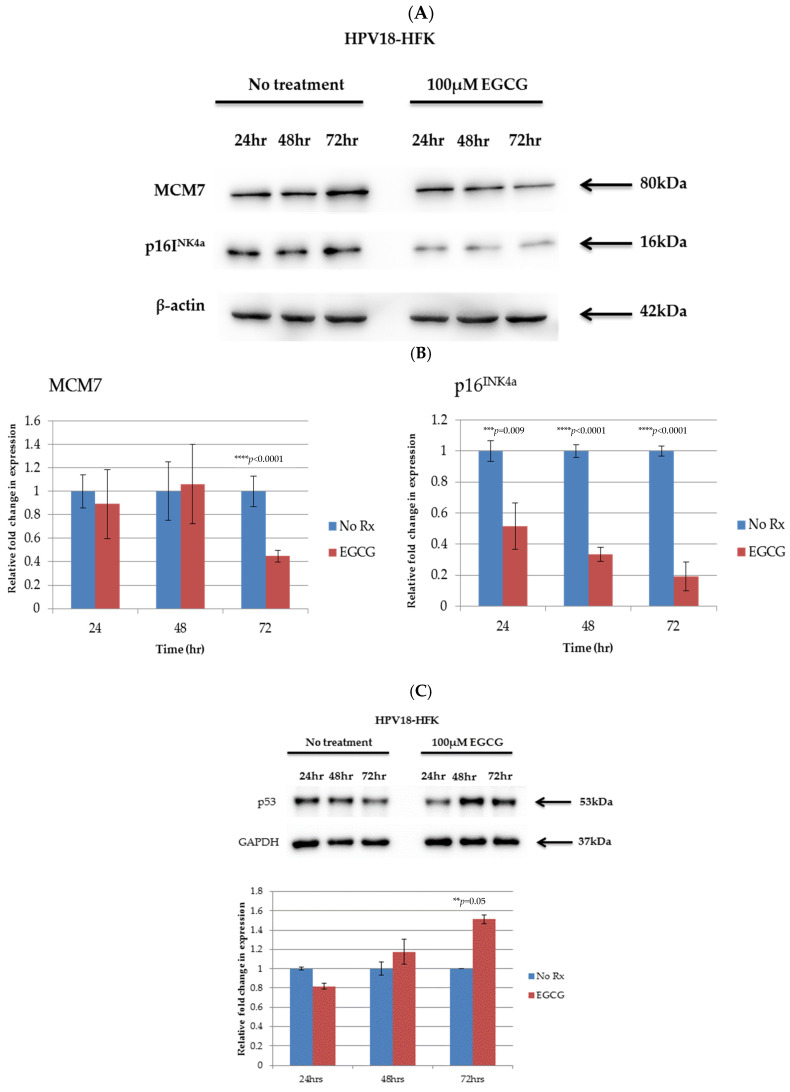
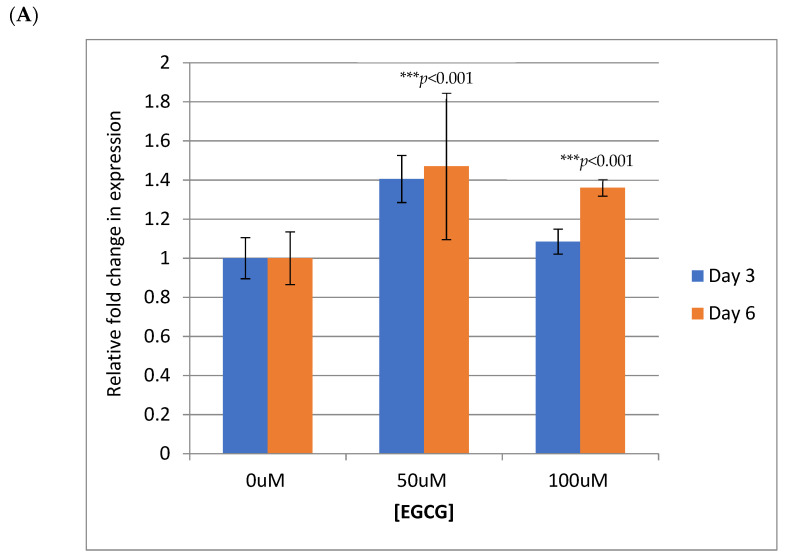
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
