

Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC
- PMID: 33920670
- PMCID: PMC8073183
- DOI: 10.3390/ijms22084173
Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC
Abstract
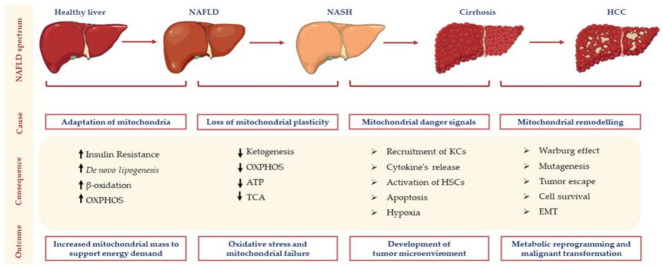
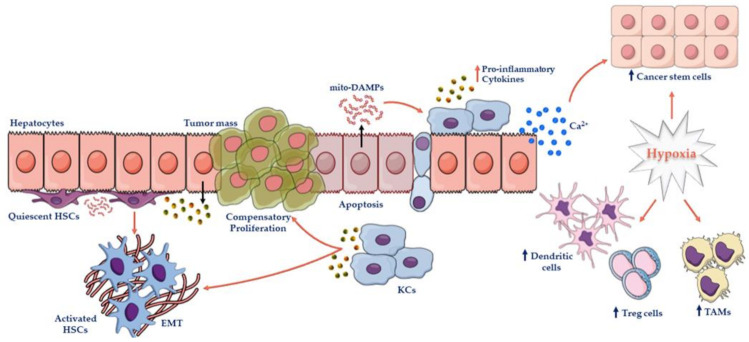
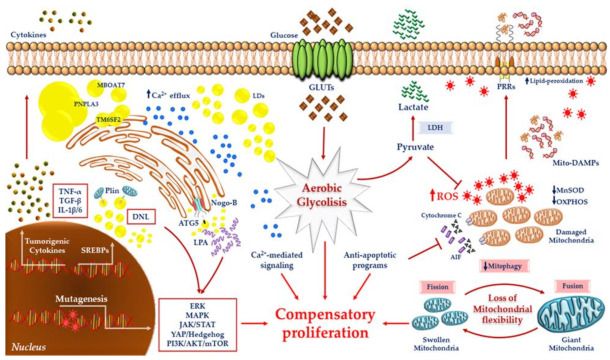
Hepatocellular carcinoma (HCC) is the most common primary malignancy of the liver and the third-leading cause of cancer-related mortality. Currently, the global burden of nonalcoholic fatty liver disease (NAFLD) has dramatically overcome both viral and alcohol hepatitis, thus becoming the main cause of HCC incidence. NAFLD pathogenesis is severely influenced by lifestyle and genetic predisposition. Mitochondria are highly dynamic organelles that may adapt in response to environment, genetics and epigenetics in the liver ("mitochondrial plasticity"). Mounting evidence highlights that mitochondrial dysfunction due to loss of mitochondrial flexibility may arise before overt NAFLD, and from the early stages of liver injury. Mitochondrial failure promotes not only hepatocellular damage, but also release signals (mito-DAMPs), which trigger inflammation and fibrosis, generating an adverse microenvironment in which several hepatocytes select anti-apoptotic programs and mutations that may allow survival and proliferation. Furthermore, one of the key events in malignant hepatocytes is represented by the remodeling of glucidic-lipidic metabolism combined with the reprogramming of mitochondrial functions, optimized to deal with energy demand. In sum, this review will discuss how mitochondrial defects may be translated into causative explanations of NAFLD-driven HCC, emphasizing future directions for research and for the development of potential preventive or curative strategies.
Keywords: HCC; HSCs; KCs; NAFLD; NASH; Warburg effect; apoptosis; hepatocytes; metabolic reprogramming; mitochondrial dynamics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Stepanova M., Rafiq N., Makhlouf H., Agrawal R., Kaur I., Younoszai Z., McCullough A., Goodman Z., Younossi Z.M. Predictors of All-Cause Mortality and Liver-Related Mortality in Patients with Non-Alcoholic Fatty Liver Disease (NAFLD) Dig. Dis. Sci. 2013;58:3017–3023. doi: 10.1007/s10620-013-2743-5. - DOI - PubMed
-
- Farrell A., Ryan M., Howell J. Epidemiology of non-alcoholic fatty liver disease-related hepatocellular carcinoma: A western perspective. Hepatoma Res. 2020;6:18. doi: 10.20517/2394-5079.2019.019. - DOI
-
- Mittal S., El-Serag H.B., Sada Y.H., Kanwal F., Duan Z., Temple S., May S.B., Kramer J.R., Richardson P.A., Davila J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2016;14:124–131.e1. doi: 10.1016/j.cgh.2015.07.019. - DOI - PMC - PubMed
-
- Bugianesi E., Leone N., Vanni E., Marchesini G., Brunello F., Carucci P., Musso A., De Paolis P., Capussotti L., Salizzoni M., et al. Expanding the natural history of nonalcoholic steatohepatitis: From cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–140. doi: 10.1053/gast.2002.34168. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
