

Genome Editing in Bacteria: CRISPR-Cas and Beyond
- PMID: 33920749
- PMCID: PMC8071187
- DOI: 10.3390/microorganisms9040844
Genome Editing in Bacteria: CRISPR-Cas and Beyond
Abstract
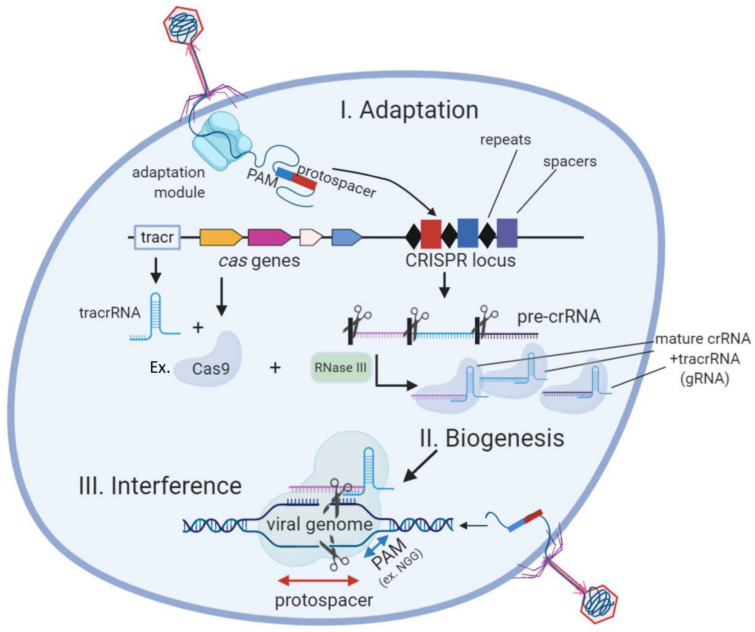
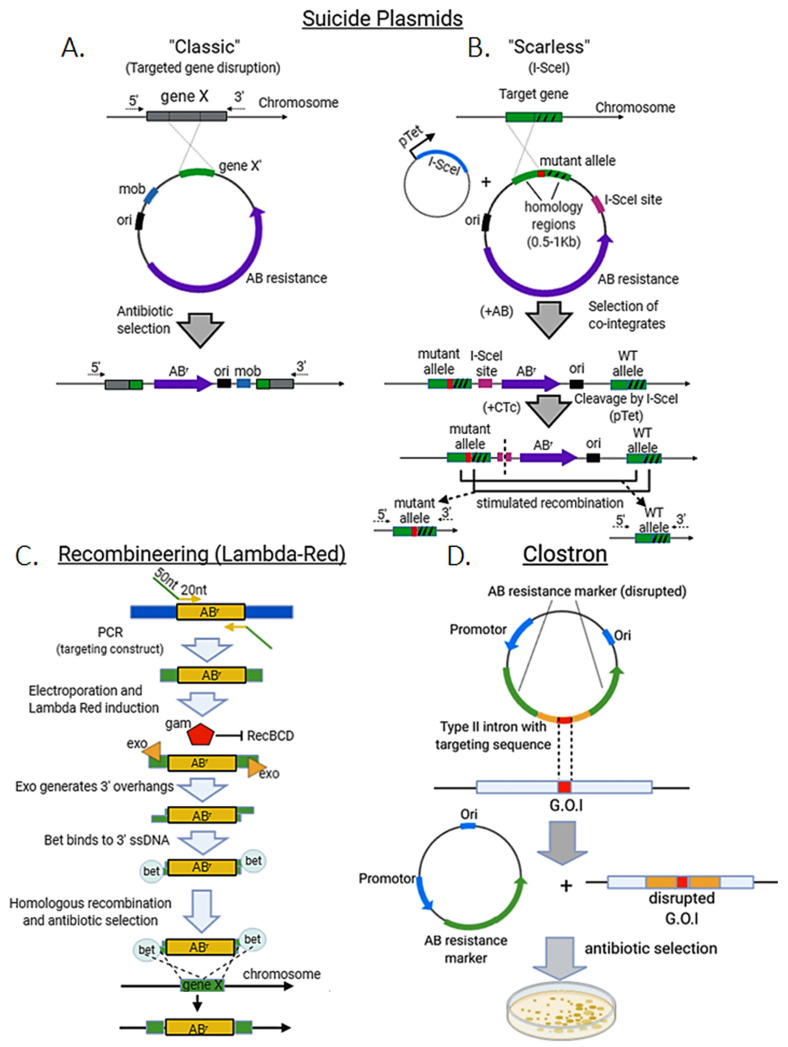
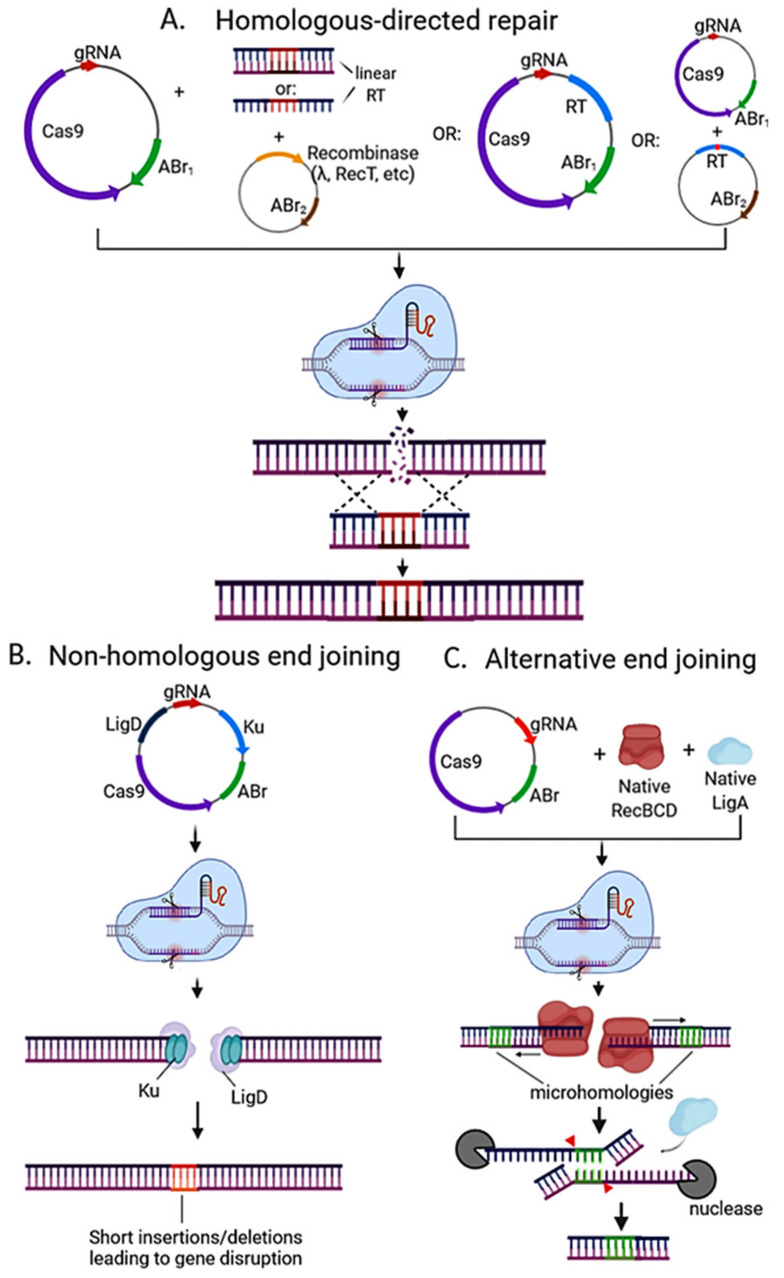
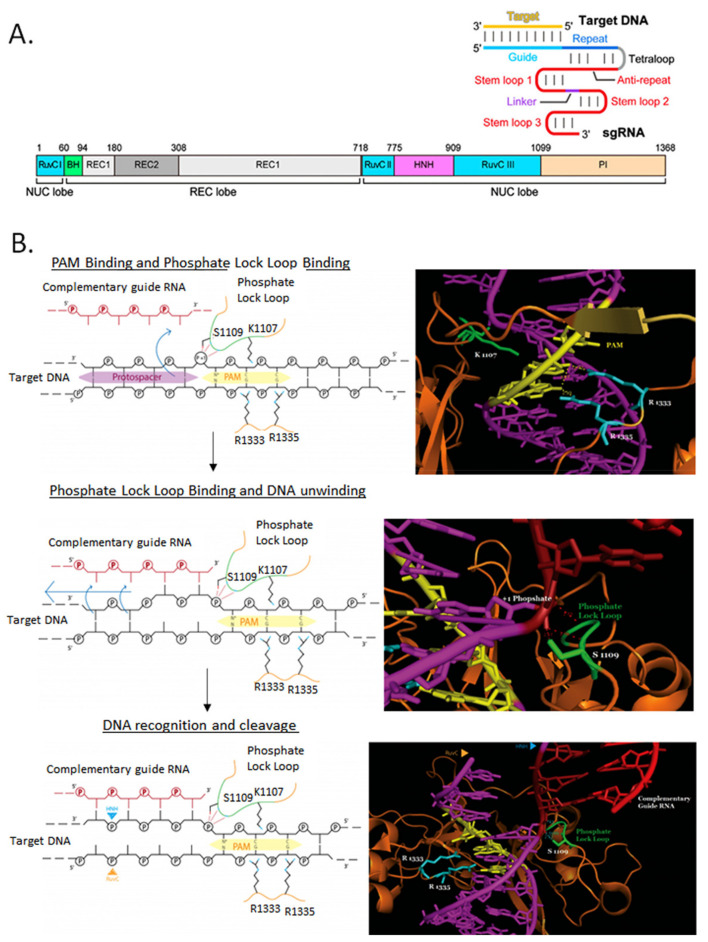
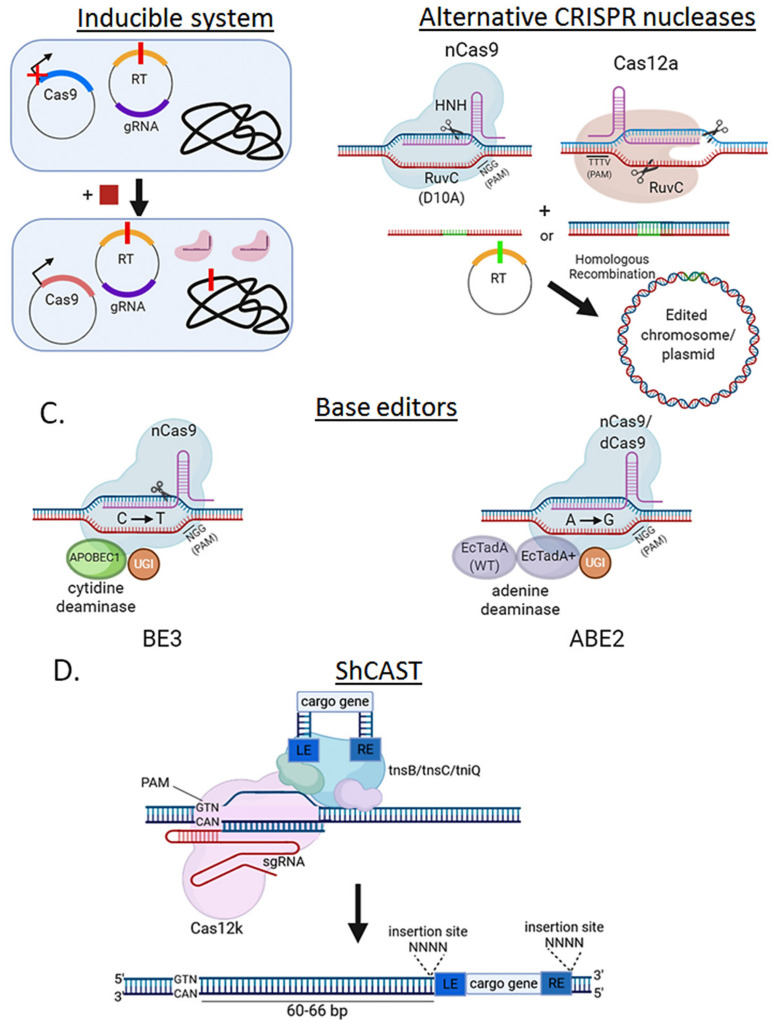
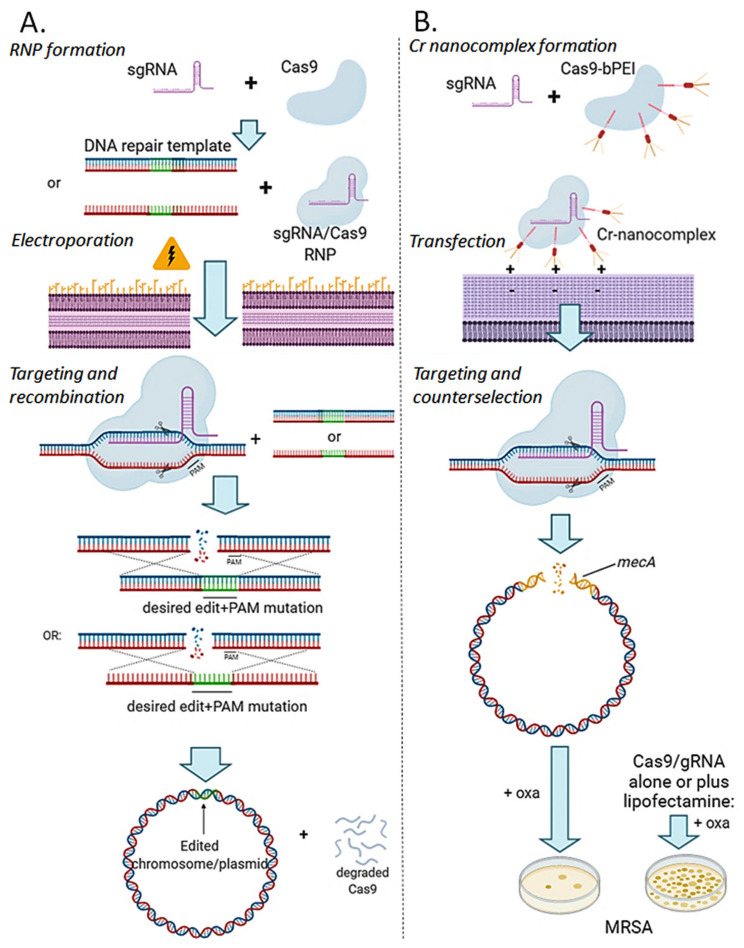
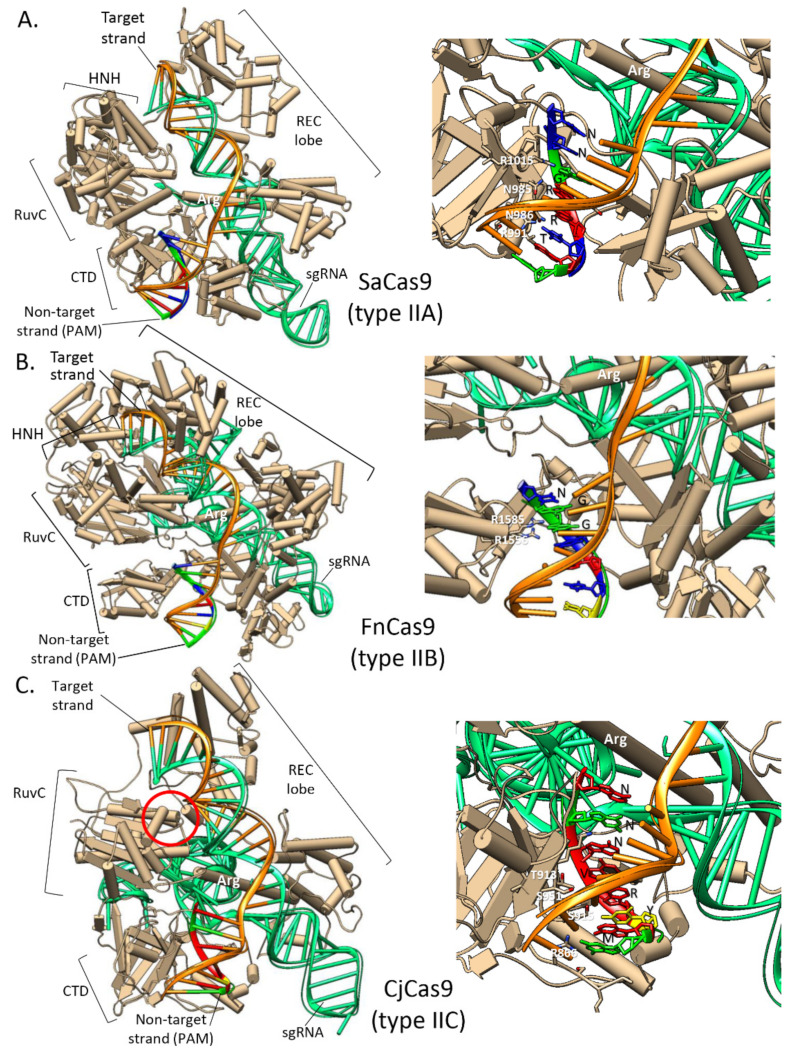
Genome editing in bacteria encompasses a wide array of laborious and multi-step methods such as suicide plasmids. The discovery and applications of clustered regularly interspaced short palindromic repeats (CRISPR)-Cas based technologies have revolutionized genome editing in eukaryotic organisms due to its simplicity and programmability. Nevertheless, this system has not been as widely favored for bacterial genome editing. In this review, we summarize the main approaches and difficulties associated with CRISPR-Cas-mediated genome editing in bacteria and present some alternatives to circumvent these issues, including CRISPR nickases, Cas12a, base editors, CRISPR-associated transposases, prime-editing, endogenous CRISPR systems, and the use of pre-made ribonucleoprotein complexes of Cas proteins and guide RNAs. Finally, we also address fluorescent-protein-based methods to evaluate the efficacy of CRISPR-based systems for genome editing in bacteria. CRISPR-Cas still holds promise as a generalized genome-editing tool in bacteria and is developing further optimization for an expanded application in these organisms. This review provides a rarely offered comprehensive view of genome editing. It also aims to familiarize the microbiology community with an ever-growing genome-editing toolbox for bacteria.
Keywords: CRISPR-Cas; genome editing; prokaryotes; ribonucleoprotein; suicide plasmids.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
