

Fabry Disease and the Heart: A Comprehensive Review
- PMID: 33922740
- PMCID: PMC8123068
- DOI: 10.3390/ijms22094434
Fabry Disease and the Heart: A Comprehensive Review
Abstract
Fabry disease (FD) is an X-linked lysosomal storage disorder caused by mutations of the GLA gene that result in a deficiency of the enzymatic activity of α-galactosidase A and consequent accumulation of glycosphingolipids in body fluids and lysosomes of the cells throughout the body. GB3 accumulation occurs in virtually all cardiac cells (cardiomyocytes, conduction system cells, fibroblasts, and endothelial and smooth muscle vascular cells), ultimately leading to ventricular hypertrophy and fibrosis, heart failure, valve disease, angina, dysrhythmias, cardiac conduction abnormalities, and sudden death. Despite available therapies and supportive treatment, cardiac involvement carries a major prognostic impact, representing the main cause of death in FD. In the last years, knowledge has substantially evolved on the pathophysiological mechanisms leading to cardiac damage, the natural history of cardiac manifestations, the late-onset phenotypes with predominant cardiac involvement, the early markers of cardiac damage, the role of multimodality cardiac imaging on the diagnosis, management and follow-up of Fabry patients, and the cardiac efficacy of available therapies. Herein, we provide a comprehensive and integrated review on the cardiac involvement of FD, at the pathophysiological, anatomopathological, laboratory, imaging, and clinical levels, as well as on the diagnosis and management of cardiac manifestations, their supportive treatment, and the cardiac efficacy of specific therapies, such as enzyme replacement therapy and migalastat.
Keywords: Fabry disease; cardiomyopathy; enzyme replacement therapy; heart; migalastat.
Conflict of interest statement
O.A. and G.M.-M. have received educational/research grants from Shire Human Genetic Therapies/Takeda and travel/accommodation support for conferences from Shire Human Genetic Therapies/Takeda, Amicus and Sanofi Genzyme. M.F.G. has received travel/accommodation support for conferences from Shire Human Genetic Therapies, Amicus, and Sanofi Genzyme. C.F. has received travel/accommodation support for conferences from Shire Human Genetic Therapies/Takeda, Amicus, and Sanofi Genzyme. The remaining authors declare no conflicts of interest.
Figures
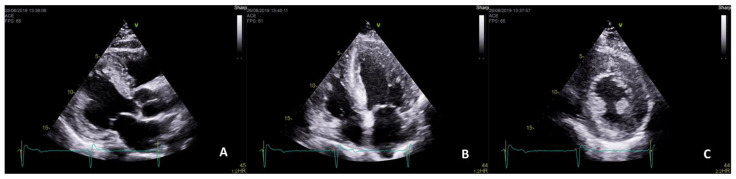
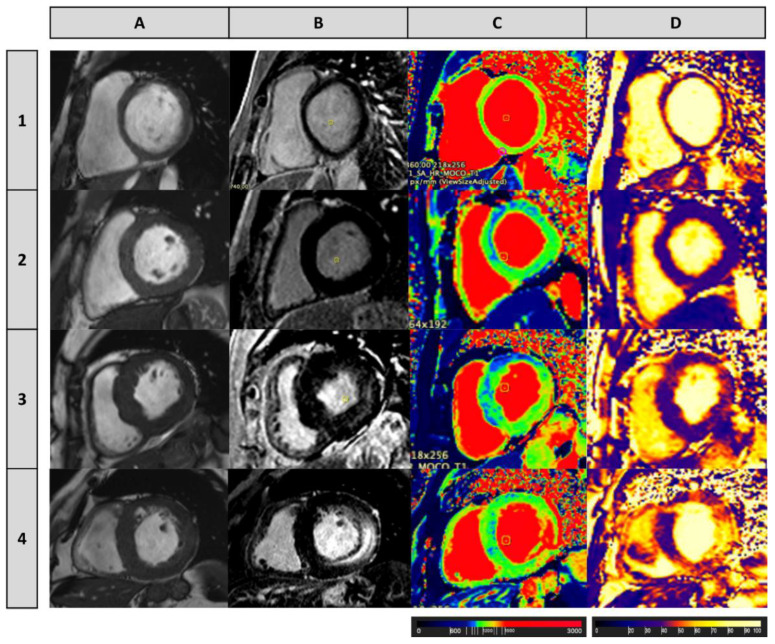
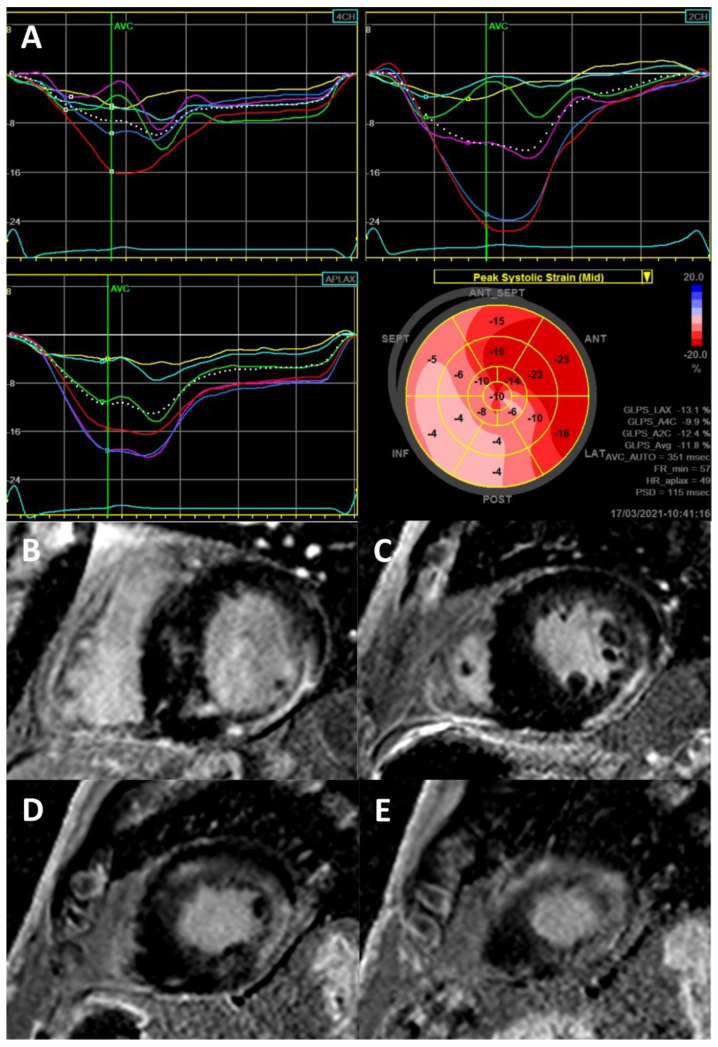
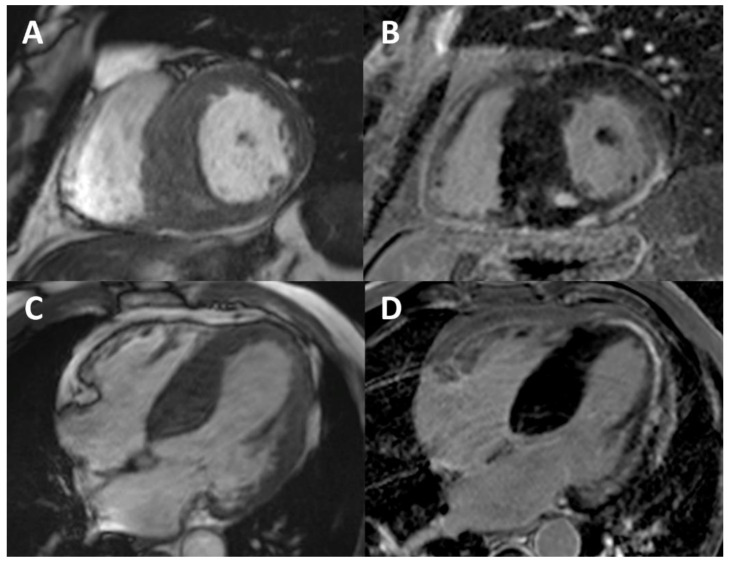
References
-
- Desnick R.J., Ioannou Y.A., Eng C.M. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., Childs B., Kinzler K.W., Vogelstein B., editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw Hill; New York, NY, USA: 2001. pp. 3733–3774.
-
- Arends M., Wanner C., Hughes D., Mehta A., Oder D., Watkinson O.T., Elliott P.M., Linthorst G.E., Wijburg F.A., Biegstraaten M., et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017;28:1631–1641. doi: 10.1681/ASN.2016090964. - DOI - PMC - PubMed
-
- Azevedo O., Gal A., Faria R., Gaspar P., Miltenberger-Miltenyi G., Gago M.F., Dias F., Martins A., Rodrigues J., Reimão P., et al. Founder effect of Fabry disease due to p.F113L mutation: Clinical profile of a late-onset phenotype. Mol. Genet. Metab. 2020;129:150–160. doi: 10.1016/j.ymgme.2019.07.012. - DOI - PubMed
-
- Azevedo O., Gago M.F., Miltenberger-Miltenyi G., Robles A.R., Costa M.A., Pereira O., Vide A.T., Branco G.C., Simões S., Guimarães M.J., et al. Natural history of the late-onset phenotype of Fabry disease due to the p.F113L mutation. Mol. Genet. Metab. Rep. 2020;22:100565. doi: 10.1016/j.ymgmr.2020.100565. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
