

An Effective MM/GBSA Protocol for Absolute Binding Free Energy Calculations: A Case Study on SARS-CoV-2 Spike Protein and the Human ACE2 Receptor
- PMID: 33923909
- PMCID: PMC8074138
- DOI: 10.3390/molecules26082383
An Effective MM/GBSA Protocol for Absolute Binding Free Energy Calculations: A Case Study on SARS-CoV-2 Spike Protein and the Human ACE2 Receptor
Abstract
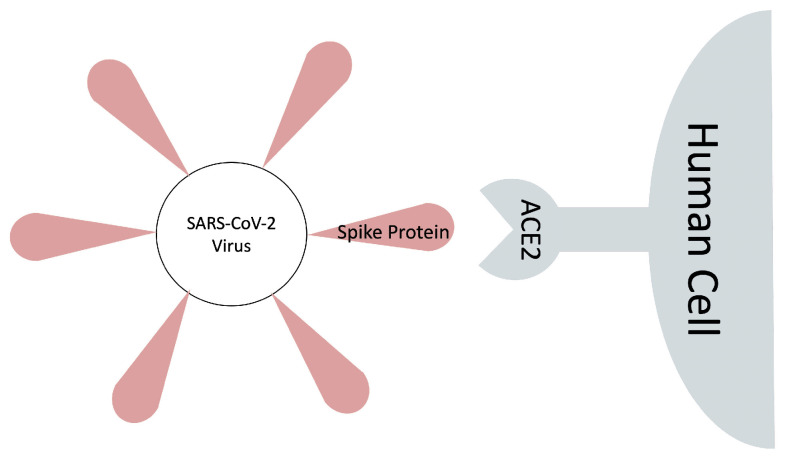
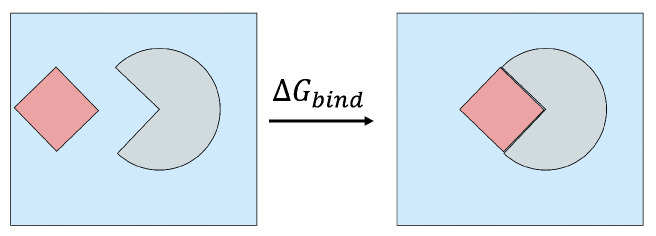
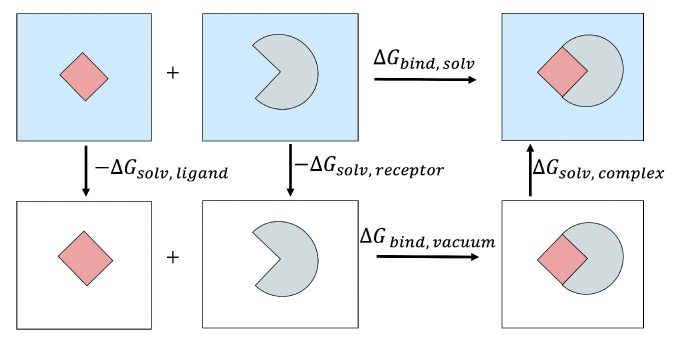
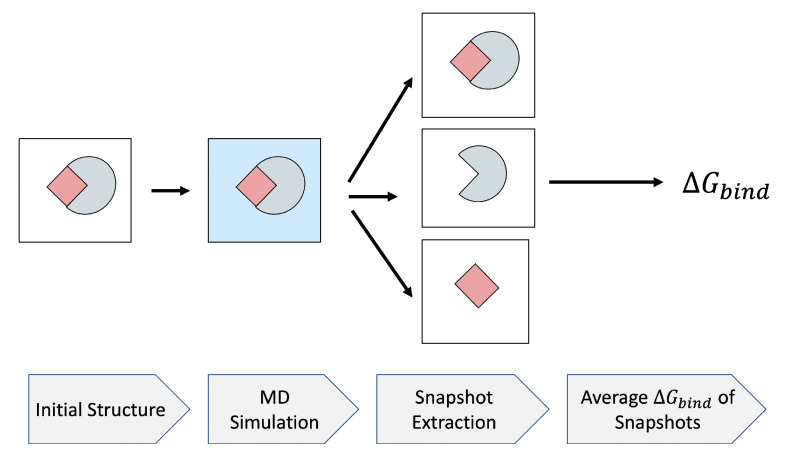
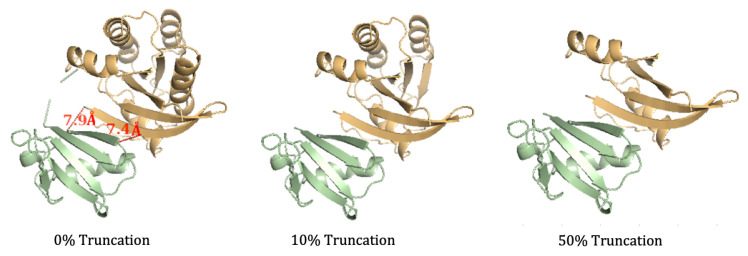
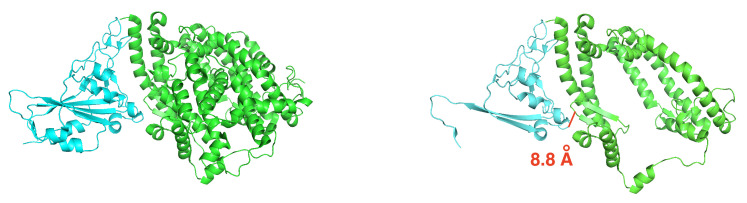
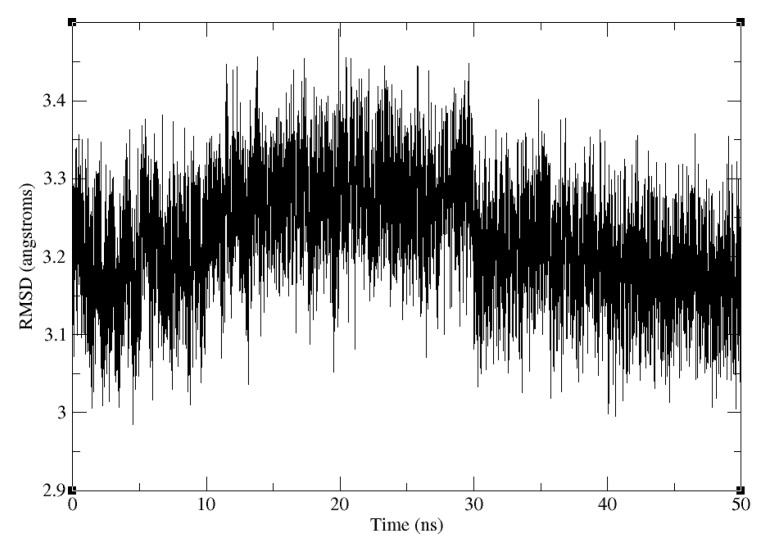
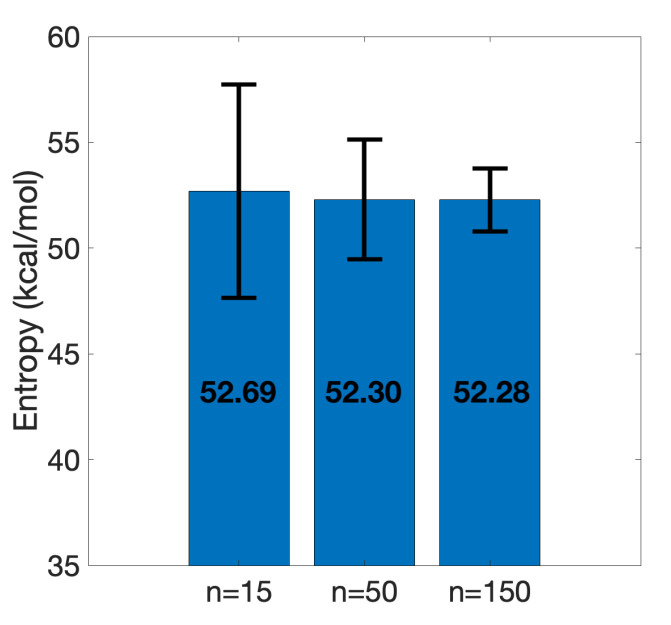
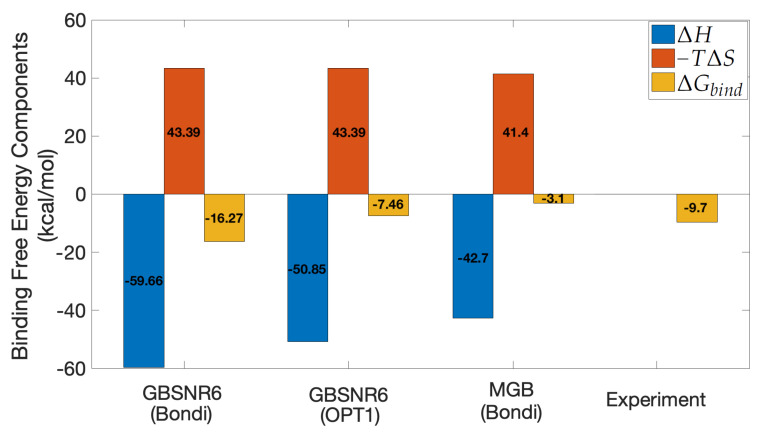
The binding free energy calculation of protein-ligand complexes is necessary for research into virus-host interactions and the relevant applications in drug discovery. However, many current computational methods of such calculations are either inefficient or inaccurate in practice. Utilizing implicit solvent models in the molecular mechanics generalized Born surface area (MM/GBSA) framework allows for efficient calculations without significant loss of accuracy. Here, GBNSR6, a new flavor of the generalized Born model, is employed in the MM/GBSA framework for measuring the binding affinity between SARS-CoV-2 spike protein and the human ACE2 receptor. A computational protocol is developed based on the widely studied Ras-Raf complex, which has similar binding free energy to SARS-CoV-2/ACE2. Two options for representing the dielectric boundary of the complexes are evaluated: one based on the standard Bondi radii and the other based on a newly developed set of atomic radii (OPT1), optimized specifically for protein-ligand binding. Predictions based on the two radii sets provide upper and lower bounds on the experimental references: -14.7(ΔGbindBondi)<-10.6(ΔGbindExp.)<-4.1(ΔGbindOPT1) kcal/mol. The consensus estimates of the two bounds show quantitative agreement with the experiment values. This work also presents a novel truncation method and computational strategies for efficient entropy calculations with normal mode analysis. Interestingly, it is observed that a significant decrease in the number of snapshots does not affect the accuracy of entropy calculation, while it does lower computation time appreciably. The proposed MM/GBSA protocol can be used to study the binding mechanism of new variants of SARS-CoV-2, as well as other relevant structures.
Keywords: SARS-CoV-2; binding free energy; entropy; implicit solvent.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
