

Small Hsps as Therapeutic Targets of Cystic Fibrosis Transmembrane Conductance Regulator Protein
- PMID: 33923911
- PMCID: PMC8072646
- DOI: 10.3390/ijms22084252
Small Hsps as Therapeutic Targets of Cystic Fibrosis Transmembrane Conductance Regulator Protein
Abstract
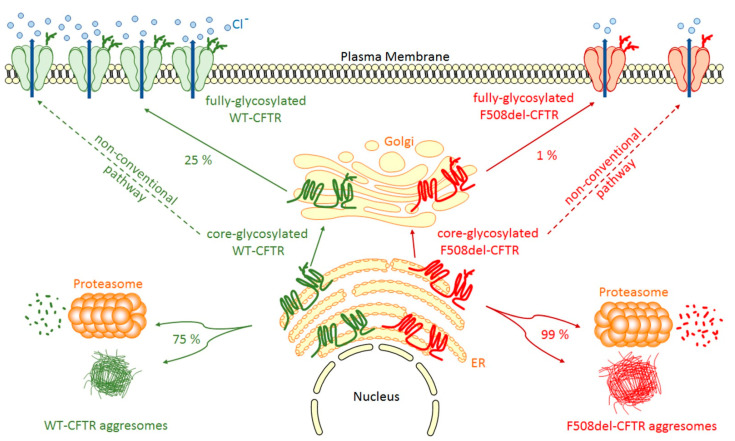
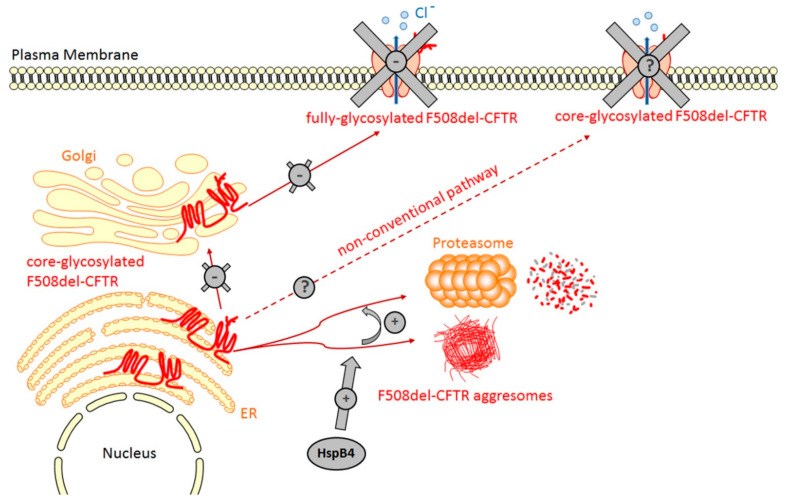
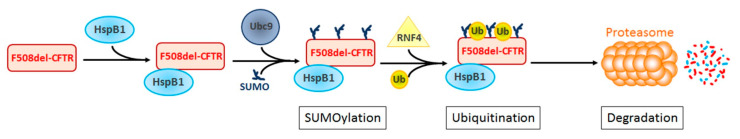
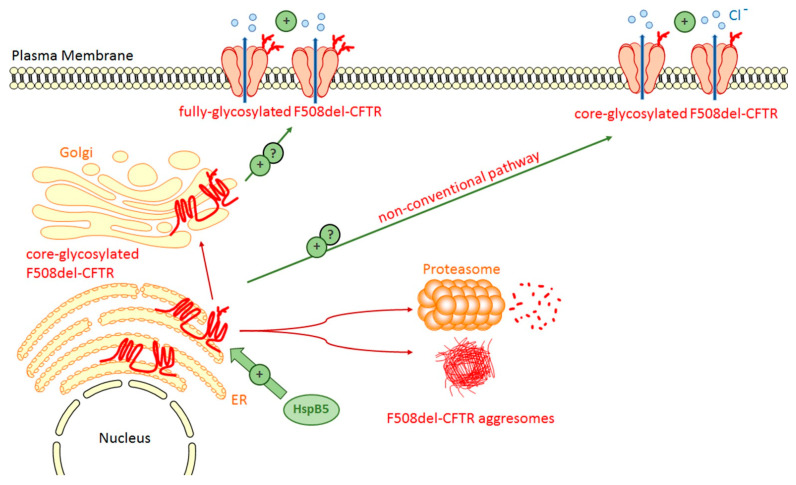
Human small heat shock proteins are molecular chaperones that regulate fundamental cellular processes in normal and pathological cells. Here, we have reviewed the role played by HspB1, HspB4 and HspB5 in the context of Cystic Fibrosis (CF), a severe monogenic autosomal recessive disease linked to mutations in Cystic Fibrosis Transmembrane conductance Regulator protein (CFTR) some of which trigger its misfolding and rapid degradation, particularly the most frequent one, F508del-CFTR. While HspB1 and HspB4 favor the degradation of CFTR mutants, HspB5 and particularly one of its phosphorylated forms positively enhance the transport at the plasma membrane, stability and function of the CFTR mutant. Moreover, HspB5 molecules stimulate the cellular efficiency of currently used CF therapeutic molecules. Different strategies are suggested to modulate the level of expression or the activity of these small heat shock proteins in view of potential in vivo therapeutic approaches. We then conclude with other small heat shock proteins that should be tested or further studied to improve our knowledge of CFTR processing.
Keywords: CFTR; HspB1; HspB4; HspB5; cystic fibrosis; sHsps.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hyde S.C., Emsley P., Hartshorn M.J., Mimmack M.M., Gileadi U., Pearce S.R., Gallagher M.P., Gill D.R., Hubbard R.E., Higgins C.F. Structural Model of ATP-Binding Proteins Associated with Cystic Fibrosis, Multidrug Resistance and Bacterial Transport. Nature. 1990;346:362–365. doi: 10.1038/346362a0. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
