

The Role of p53 Signaling in Colorectal Cancer
- PMID: 33924934
- PMCID: PMC8125348
- DOI: 10.3390/cancers13092125
The Role of p53 Signaling in Colorectal Cancer
Abstract
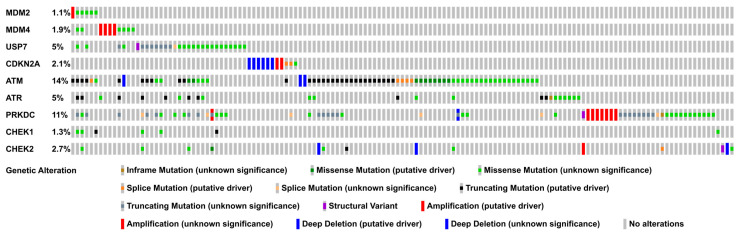
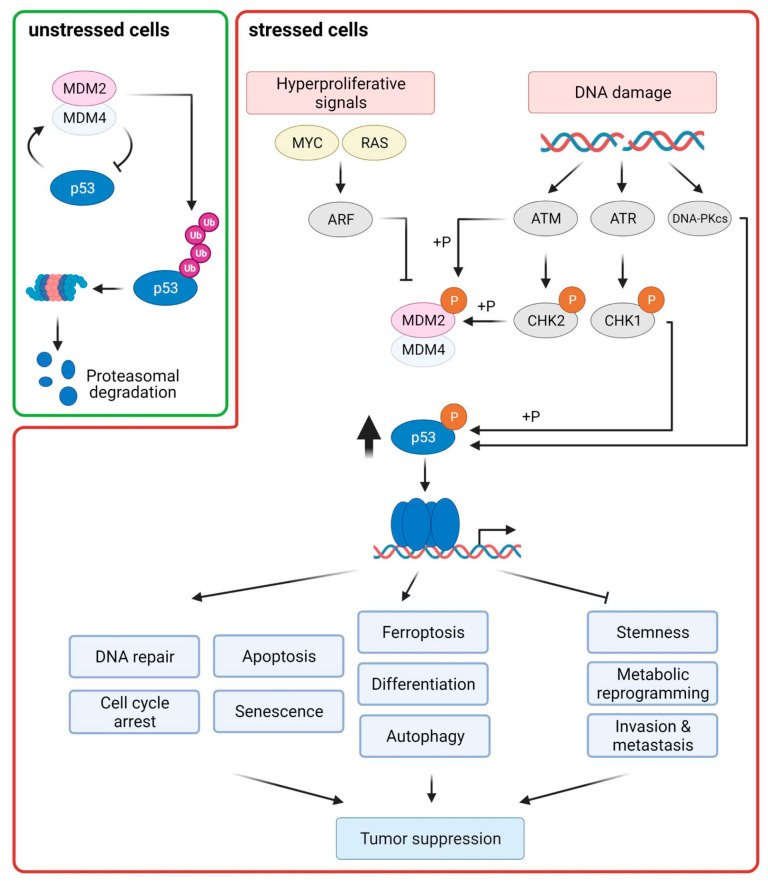
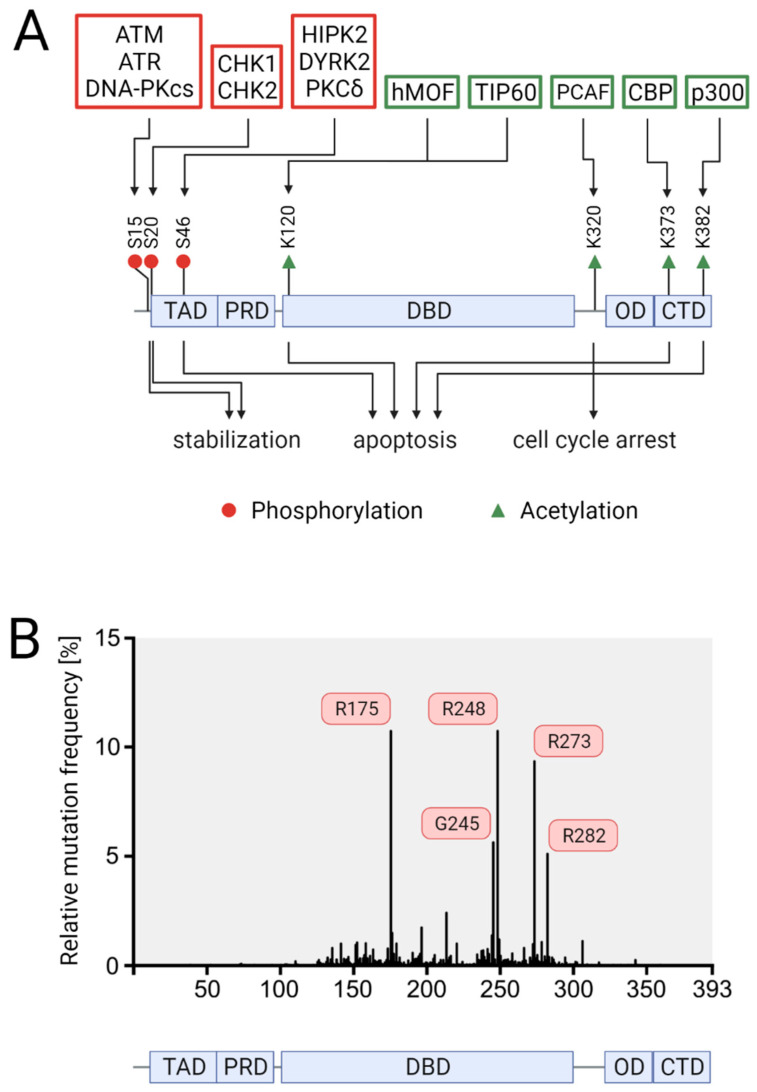
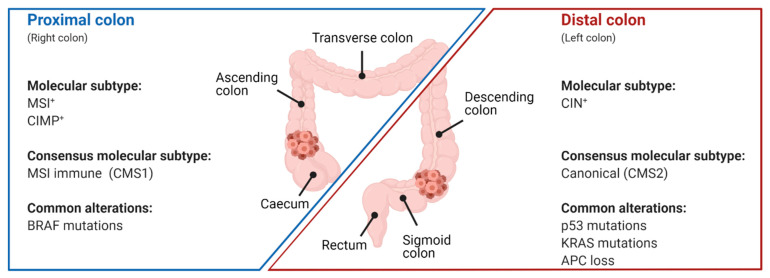
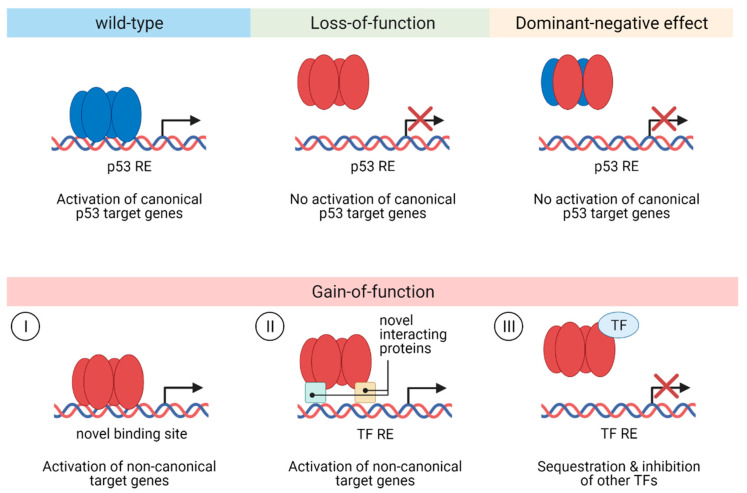
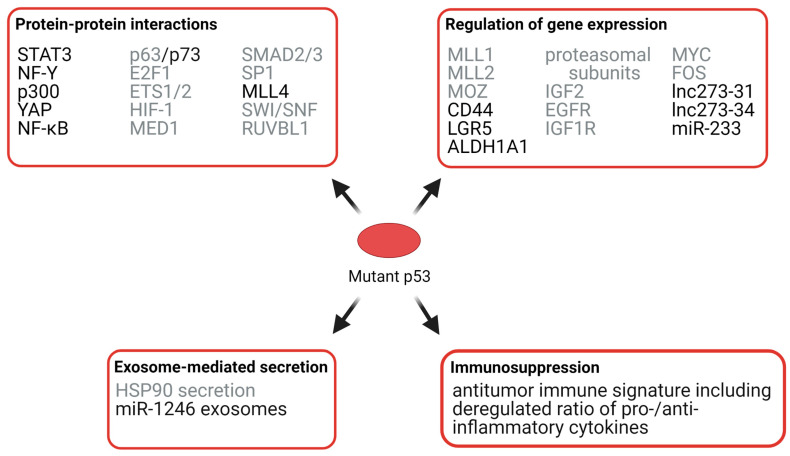
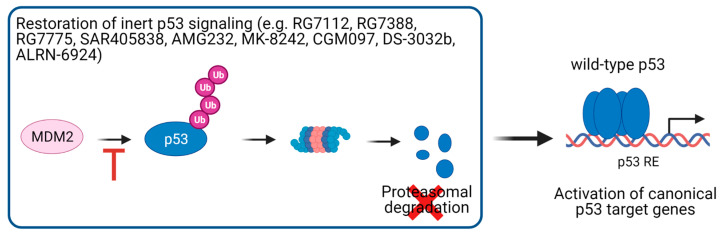
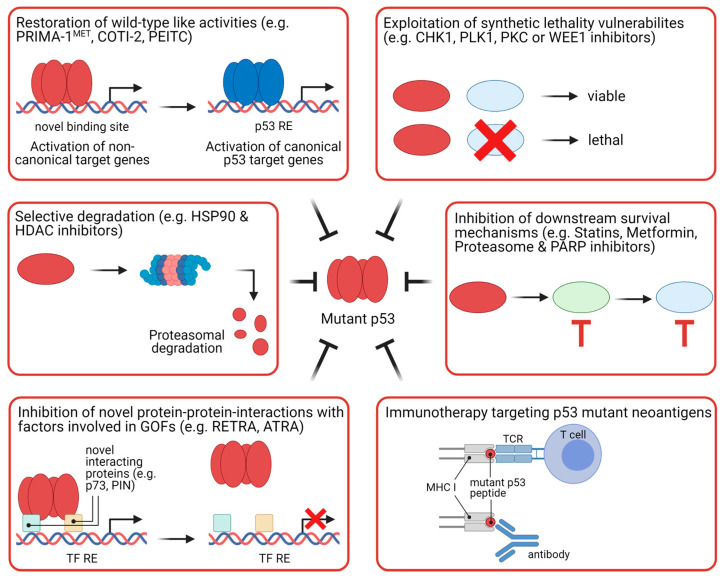
The transcription factor p53 functions as a critical tumor suppressor by orchestrating a plethora of cellular responses such as DNA repair, cell cycle arrest, cellular senescence, cell death, cell differentiation, and metabolism. In unstressed cells, p53 levels are kept low due to its polyubiquitination by the E3 ubiquitin ligase MDM2. In response to various stress signals, including DNA damage and aberrant growth signals, the interaction between p53 and MDM2 is blocked and p53 becomes stabilized, allowing p53 to regulate a diverse set of cellular responses mainly through the transactivation of its target genes. The outcome of p53 activation is controlled by its dynamics, its interactions with other proteins, and post-translational modifications. Due to its involvement in several tumor-suppressing pathways, p53 function is frequently impaired in human cancers. In colorectal cancer (CRC), the TP53 gene is mutated in 43% of tumors, and the remaining tumors often have compromised p53 functioning because of alterations in the genes encoding proteins involved in p53 regulation, such as ATM (13%) or DNA-PKcs (11%). TP53 mutations in CRC are usually missense mutations that impair wild-type p53 function (loss-of-function) and that even might provide neo-morphic (gain-of-function) activities such as promoting cancer cell stemness, cell proliferation, invasion, and metastasis, thereby promoting cancer progression. Although the first compounds targeting p53 are in clinical trials, a better understanding of wild-type and mutant p53 functions will likely pave the way for novel CRC therapies.
Keywords: cancer therapy; colorectal cancer; gain-of-function; mutant p53; p53 pathway; p53 signaling; wild type p53.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
