

The impact of cell type and context-dependent regulatory variants on human immune traits
- PMID: 33926512
- PMCID: PMC8082814
- DOI: 10.1186/s13059-021-02334-x
The impact of cell type and context-dependent regulatory variants on human immune traits
Abstract
Background: The vast majority of trait-associated variants identified using genome-wide association studies (GWAS) are noncoding, and therefore assumed to impact gene regulation. However, the majority of trait-associated loci are unexplained by regulatory quantitative trait loci (QTLs).
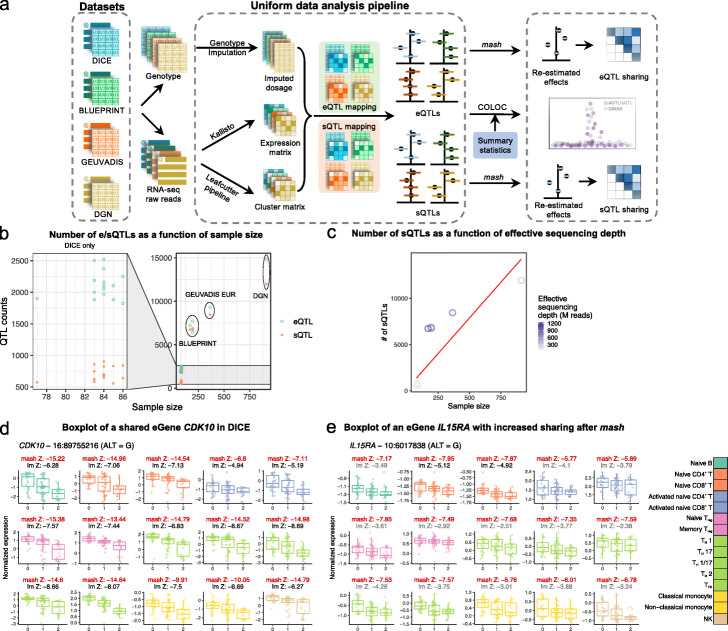
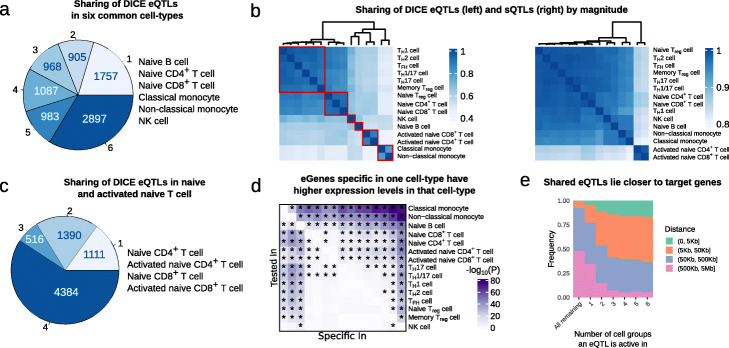
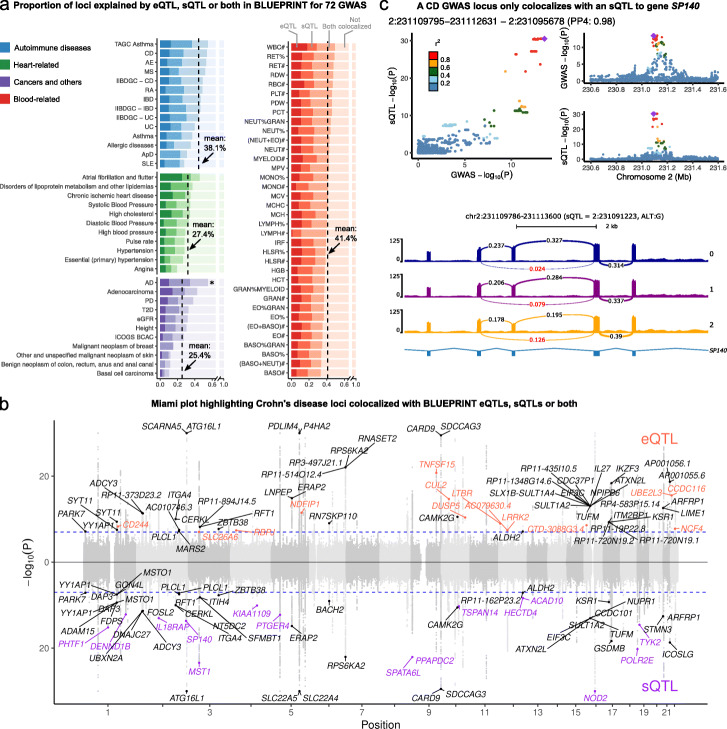
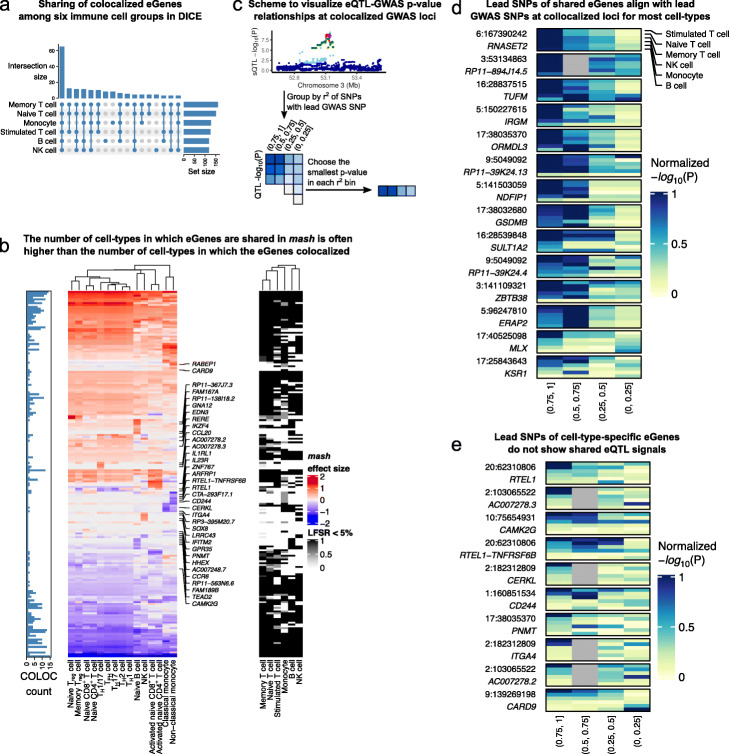
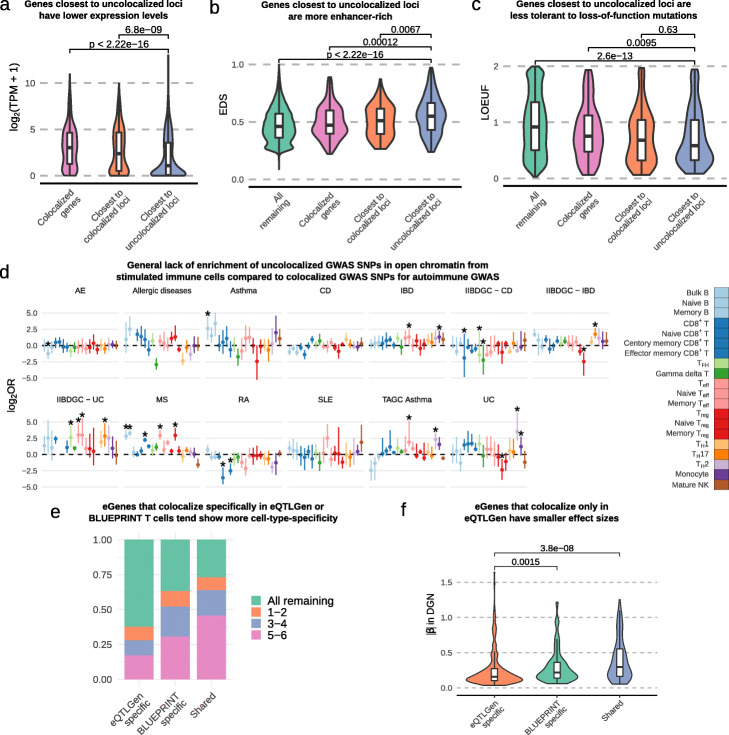
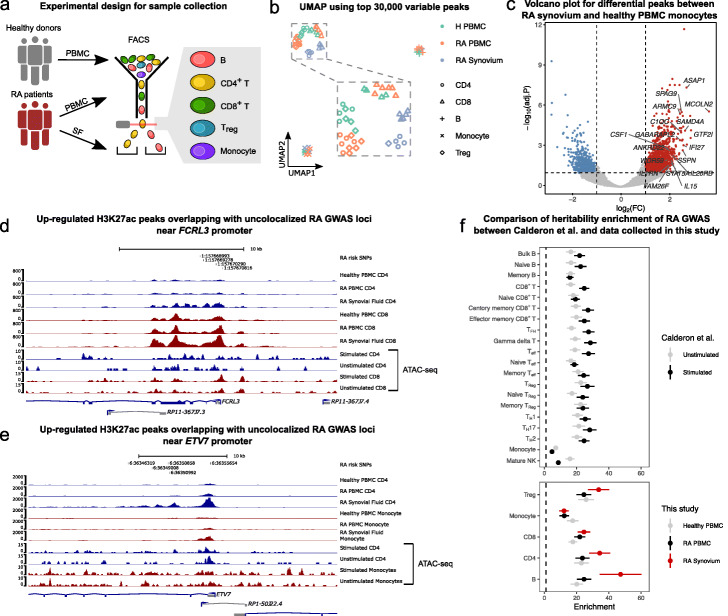
Results: We perform a comprehensive characterization of the putative mechanisms by which GWAS loci impact human immune traits. By harmonizing four major immune QTL studies, we identify 26,271 expression QTLs (eQTLs) and 23,121 splicing QTLs (sQTLs) spanning 18 immune cell types. Our colocalization analyses between QTLs and trait-associated loci from 72 GWAS reveals that genetic effects on RNA expression and splicing in immune cells colocalize with 40.4% of GWAS loci for immune-related traits, in many cases increasing the fraction of colocalized loci by two fold compared to previous studies. Notably, we find that the largest contributors of this increase are splicing QTLs, which colocalize on average with 14% of all GWAS loci that do not colocalize with eQTLs. By contrast, we find that cell type-specific eQTLs, and eQTLs with small effect sizes contribute very few new colocalizations. To investigate the 60% of GWAS loci that remain unexplained, we collect H3K27ac CUT&Tag data from rheumatoid arthritis and healthy controls, and find large-scale differences between immune cells from the different disease contexts, including at regions overlapping unexplained GWAS loci.
Conclusion: Altogether, our work supports RNA splicing as an important mediator of genetic effects on immune traits, and suggests that we must expand our study of regulatory processes in disease contexts to improve functional interpretation of as yet unexplained GWAS loci.
Keywords: Colocalization; GWAS; eQTL; sQTL.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Subcutaneous adipose tissue splice quantitative trait loci reveal differences in isoform usage associated with cardiometabolic traits.Am J Hum Genet. 2022 Jan 6;109(1):66-80. doi: 10.1016/j.ajhg.2021.11.019. Am J Hum Genet. 2022. PMID: 34995504 Free PMC article.
-
Multiomic QTL mapping reveals phenotypic complexity of GWAS loci and prioritizes putative causal variants.Cell Genom. 2025 Mar 12;5(3):100775. doi: 10.1016/j.xgen.2025.100775. Epub 2025 Feb 21. Cell Genom. 2025. PMID: 39986281 Free PMC article.
-
Cell-type-specific meQTLs extend melanoma GWAS annotation beyond eQTLs and inform melanocyte gene-regulatory mechanisms.Am J Hum Genet. 2021 Sep 2;108(9):1631-1646. doi: 10.1016/j.ajhg.2021.06.018. Epub 2021 Jul 21. Am J Hum Genet. 2021. PMID: 34293285 Free PMC article.
-
QTL Analysis Beyond eQTLs.Methods Mol Biol. 2020;2082:201-210. doi: 10.1007/978-1-0716-0026-9_14. Methods Mol Biol. 2020. PMID: 31849017 Review.
-
An overview of detecting gene-trait associations by integrating GWAS summary statistics and eQTLs.Sci China Life Sci. 2024 Jun;67(6):1133-1154. doi: 10.1007/s11427-023-2522-8. Epub 2024 Mar 29. Sci China Life Sci. 2024. PMID: 38568343 Review.
Cited by
-
Functional genomics implicates natural killer cells in the pathogenesis of ankylosing spondylitis.HGG Adv. 2025 Jan 9;6(1):100375. doi: 10.1016/j.xhgg.2024.100375. Epub 2024 Oct 28. HGG Adv. 2025. PMID: 39468794 Free PMC article.
-
A scoping review of statistical methods to investigate colocalization between genetic associations and microRNA expression in osteoarthritis.Osteoarthr Cartil Open. 2024 Nov 8;6(4):100540. doi: 10.1016/j.ocarto.2024.100540. eCollection 2024 Dec. Osteoarthr Cartil Open. 2024. PMID: 39640910 Free PMC article.
-
The missing link between genetic association and regulatory function.Elife. 2022 Dec 14;11:e74970. doi: 10.7554/eLife.74970. Elife. 2022. PMID: 36515579 Free PMC article.
-
Complement component C4 structural variation and quantitative traits contribute to sex-biased vulnerability in systemic sclerosis.NPJ Genom Med. 2022 Oct 5;7(1):57. doi: 10.1038/s41525-022-00327-8. NPJ Genom Med. 2022. PMID: 36198672 Free PMC article.
-
Cell-type-specific and disease-associated expression quantitative trait loci in the human lung.Nat Genet. 2024 Apr;56(4):595-604. doi: 10.1038/s41588-024-01702-0. Epub 2024 Mar 28. Nat Genet. 2024. PMID: 38548990 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
