

Crosstalk between microRNA expression and DNA methylation drives the hormone-dependent phenotype of breast cancer
- PMID: 33926515
- PMCID: PMC8086068
- DOI: 10.1186/s13073-021-00880-4
Crosstalk between microRNA expression and DNA methylation drives the hormone-dependent phenotype of breast cancer
Abstract
Background: Abnormal DNA methylation is observed as an early event in breast carcinogenesis. However, how such alterations arise is still poorly understood. microRNAs (miRNAs) regulate gene expression at the post-transcriptional level and play key roles in various biological processes. Here, we integrate miRNA expression and DNA methylation at CpGs to study how miRNAs may affect the breast cancer methylome and how DNA methylation may regulate miRNA expression.
Methods: miRNA expression and DNA methylation data from two breast cancer cohorts, Oslo2 (n = 297) and The Cancer Genome Atlas (n = 439), were integrated through a correlation approach that we term miRNA-methylation Quantitative Trait Loci (mimQTL) analysis. Hierarchical clustering was used to identify clusters of miRNAs and CpGs that were further characterized through analysis of mRNA/protein expression, clinicopathological features, in silico deconvolution, chromatin state and accessibility, transcription factor binding, and long-range interaction data.
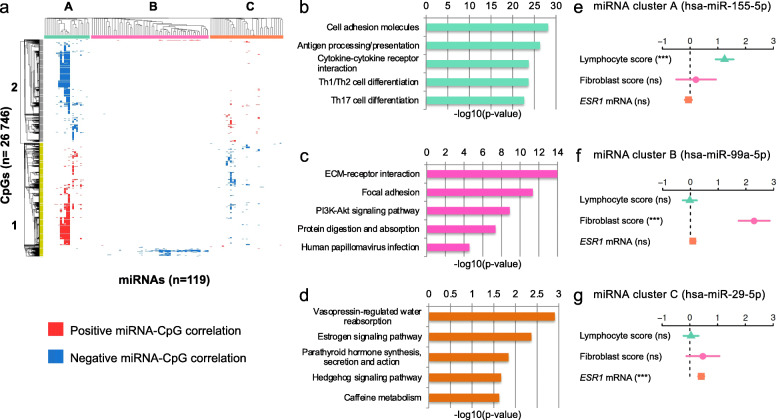
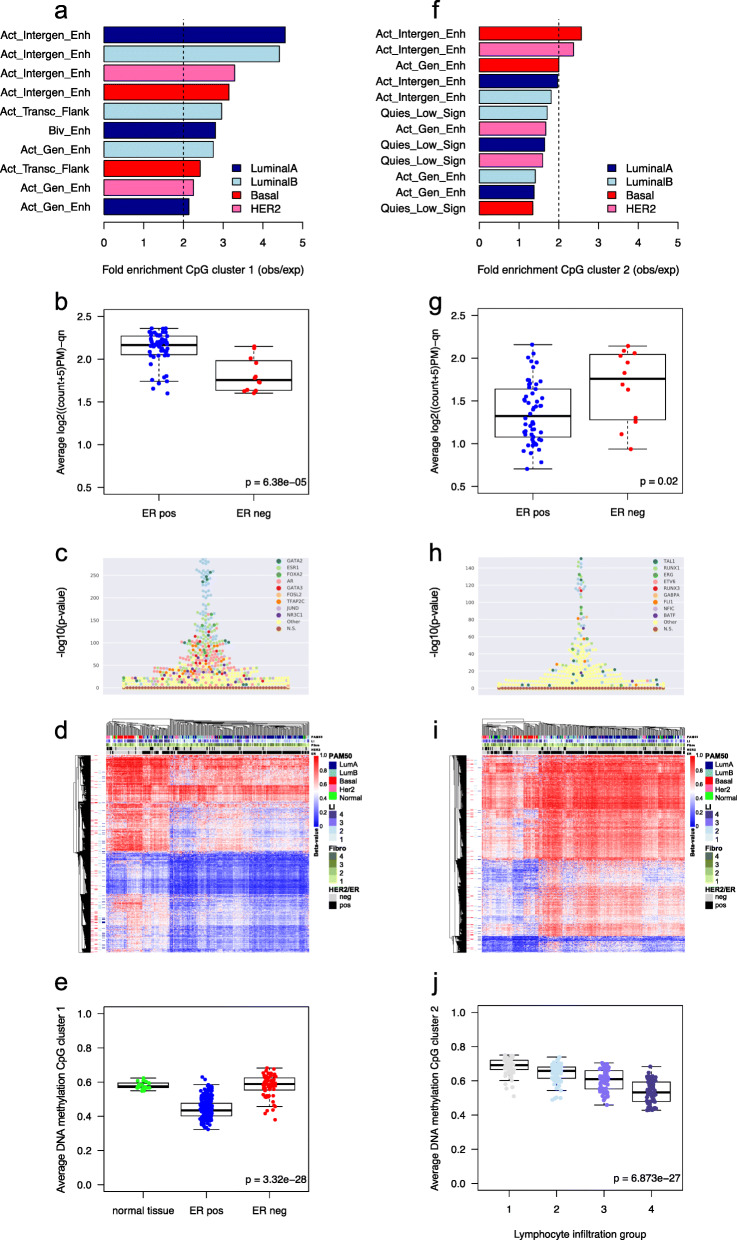
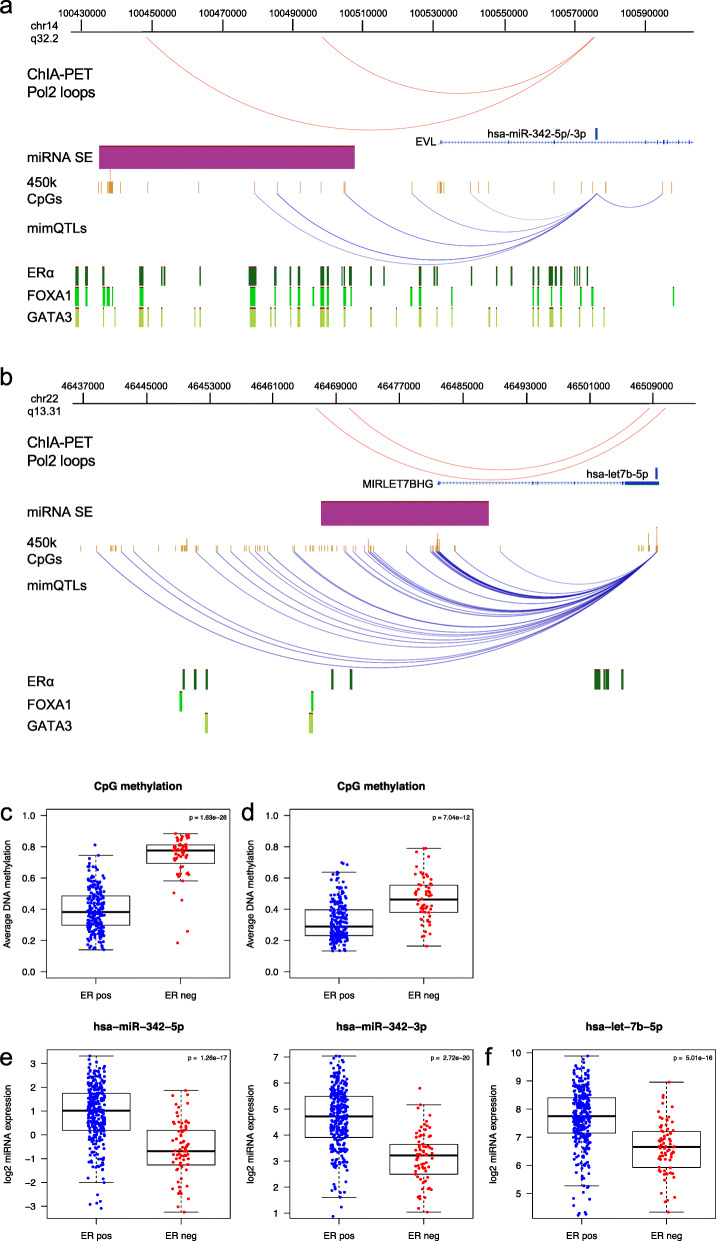
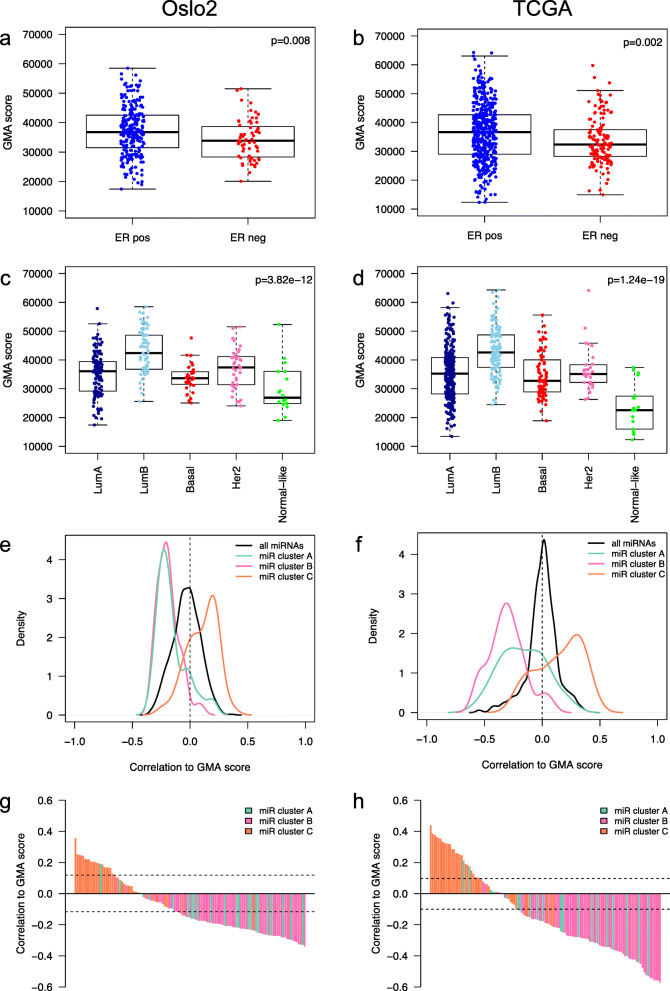
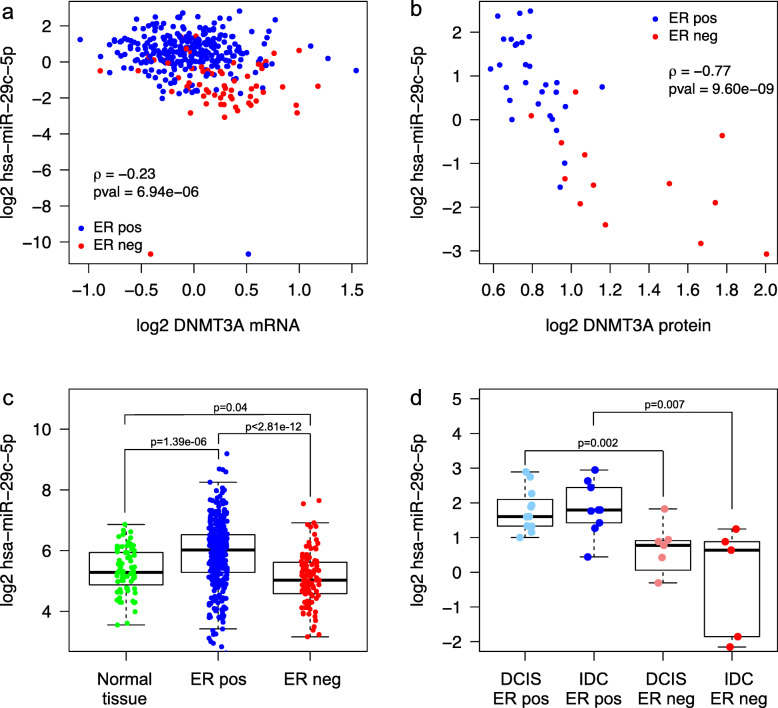
Results: Clustering of the significant mimQTLs identified distinct groups of miRNAs and CpGs that reflect important biological processes associated with breast cancer pathogenesis. Notably, two major miRNA clusters were related to immune or fibroblast infiltration, hence identifying miRNAs associated with cells of the tumor microenvironment, while another large cluster was related to estrogen receptor (ER) signaling. Studying the chromatin landscape surrounding CpGs associated with the estrogen signaling cluster, we found that miRNAs from this cluster are likely to be regulated through DNA methylation of enhancers bound by FOXA1, GATA2, and ER-alpha. Further, at the hub of the estrogen cluster, we identified hsa-miR-29c-5p as negatively correlated with the mRNA and protein expression of DNA methyltransferase DNMT3A, a key enzyme regulating DNA methylation. We found deregulation of hsa-miR-29c-5p already present in pre-invasive breast lesions and postulate that hsa-miR-29c-5p may trigger early event abnormal DNA methylation in ER-positive breast cancer.
Conclusions: We describe how miRNA expression and DNA methylation interact and associate with distinct breast cancer phenotypes.
Keywords: Breast cancer; Methylation; Omics integration; Systems biology; miRNA.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Fleischer T, Frigessi A, Johnson KC, Edvardsen H, Touleimat N, Klajic J, Riis ML, Haakensen VD, Wärnberg F, Naume B, et al. Genome-wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014;15:435. - PMC - PubMed
-
- Stefansson OA, Moran S, Gomez A, Sayols S, Arribas-Jorba C, Sandoval J, Hilmarsdottir H, Olafsdottir E, Tryggvadottir L, Jonasson JG, Eyfjord J, Esteller M. A DNA methylation-based definition of biologically distinct breast cancer subtypes. Mol Oncol. 2015;9(3):555–568. doi: 10.1016/j.molonc.2014.10.012. - DOI - PMC - PubMed
-
- Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17(3):330–9. 10.1038/nm.2305. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
