

Pulmonary granulomatosis of genetic origin
- PMID: 33927005
- PMCID: PMC9488645
- DOI: 10.1183/16000617.0152-2020
Pulmonary granulomatosis of genetic origin
Abstract
Granulomatous inflammation of the lung can be a manifestation of different conditions and can be caused by endogenous inflammation or external triggers. A multitude of different genetic mutations can either predispose patients to infections with granuloma-forming pathogens or cause autoinflammatory disorders, both leading to the phenotype of pulmonary granulomatosis. Based on a detailed patient history, physical examination and a diagnostic approach including laboratory workup, pulmonary function tests (PFTs), computed tomography (CT) scans, bronchoscopy with bronchoalveolar lavage (BAL), lung biopsies and specialised microbiological and immunological diagnostics, a correct diagnosis of an underlying cause of pulmonary granulomatosis of genetic origin can be made and appropriate therapy can be initiated. Depending on the underlying disorder, treatment approaches can include antimicrobial therapy, immunosuppression and even haematopoietic stem cell transplantation (HSCT). Patients with immunodeficiencies and autoinflammatory conditions are at the highest risk of developing pulmonary granulomatosis of genetic origin. Here we provide a review on these disorders and discuss pathogenesis, clinical presentation, diagnostic approach and treatment.
Copyright ©ERS 2021.
Conflict of interest statement
Conflict of interest: S.F.N. Bode has nothing to disclose. Conflict of interest: J. Rohr has nothing to disclose. Conflict of interest: J.M. Müller-Quernheim reports grants and non-financial support from Bristol Myers Squibb; personal fees from Roche and Novartis; personal fees and non-financial support from Boehringer Ingelheim; and non-financial support from CLS Behring, outside the submitted work. In addition, J.M. Müller-Quernheim has a patent “Use of inhaled VIP for treatment of immune checkpoint inhibitor-induced pneumonitis” pending and is co-founder of AdVita Lifescience GmbH, a spin-off of the University of Freiburg. Advita reports a pending intellectual property concerning the use of Aviptadil for the treatment of immune checkpoint inhibitor (ICI) pneumonitis. Conflict of interest: M. Seidl has nothing to disclose. Conflict of interest: C. Speckmann has nothing to disclose. Conflict of interest: A. Heinzmann has nothing to disclose.
Figures
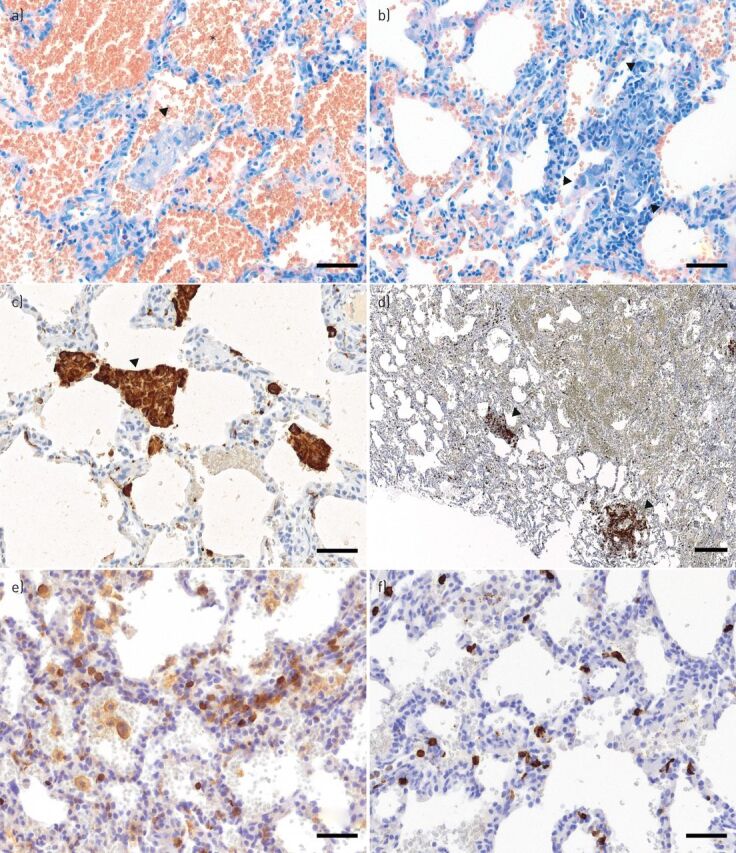
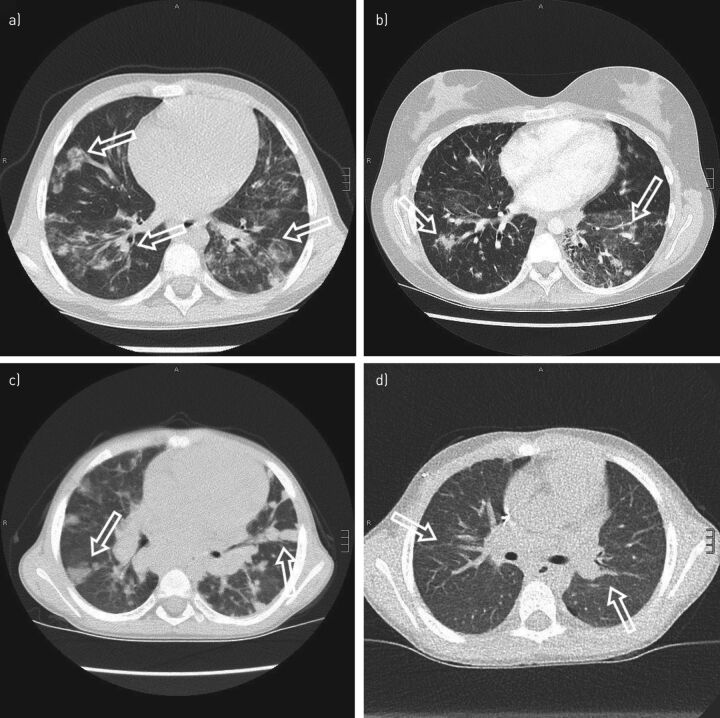
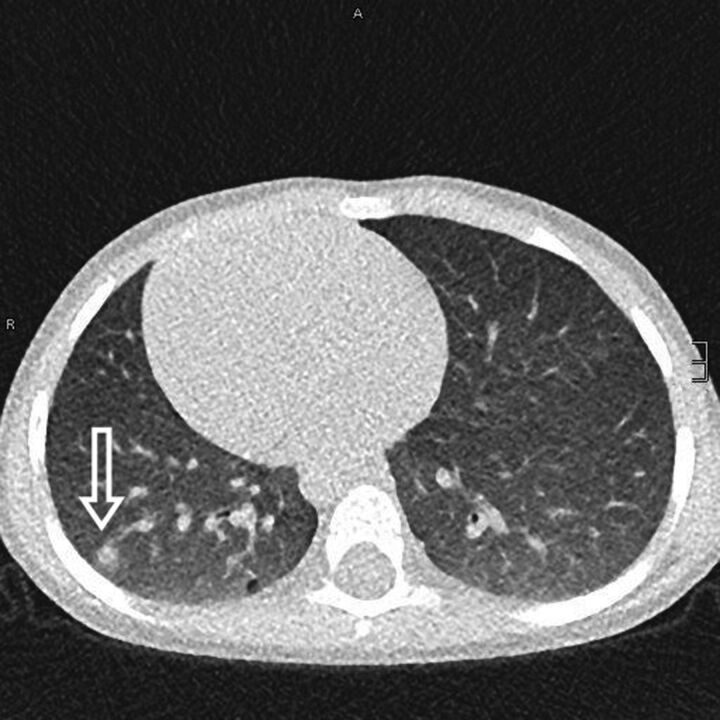
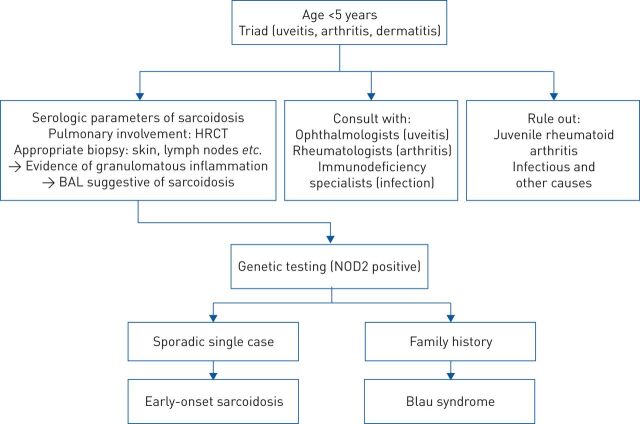
References
-
- Mukhopadhyay S, Gal AA. Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med 2010; 134: 667–690. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources