

A single-embryo, single-cell time-resolved model for mouse gastrulation
- PMID: 33932341
- PMCID: PMC8162424
- DOI: 10.1016/j.cell.2021.04.004
A single-embryo, single-cell time-resolved model for mouse gastrulation
Abstract
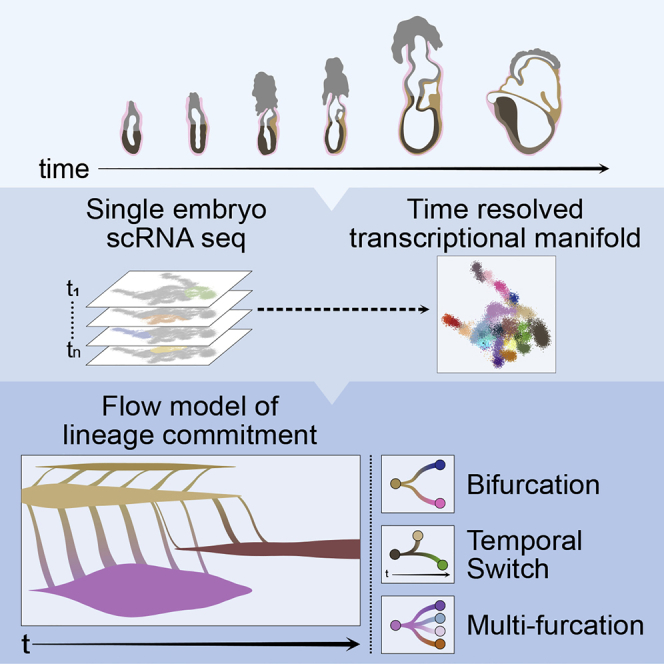
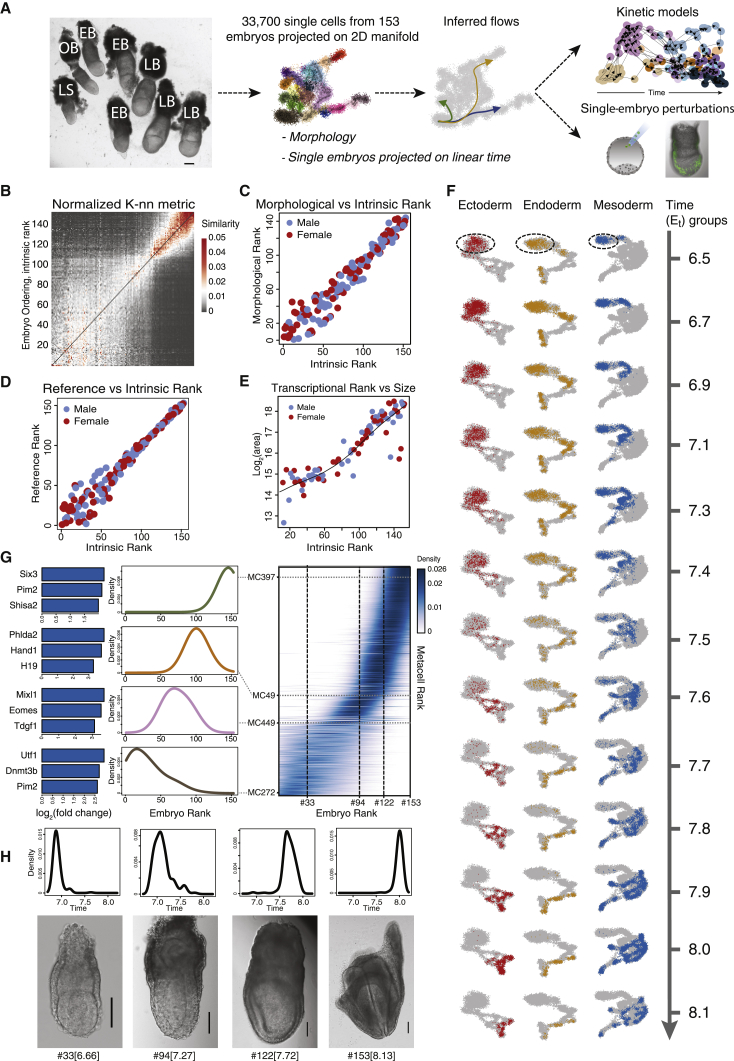
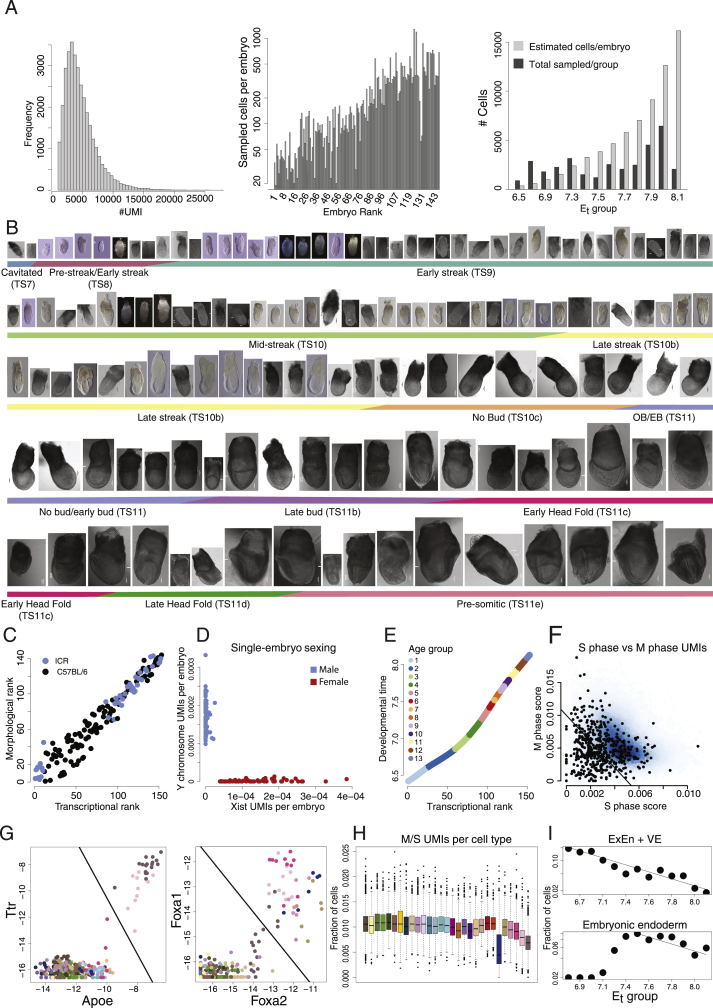
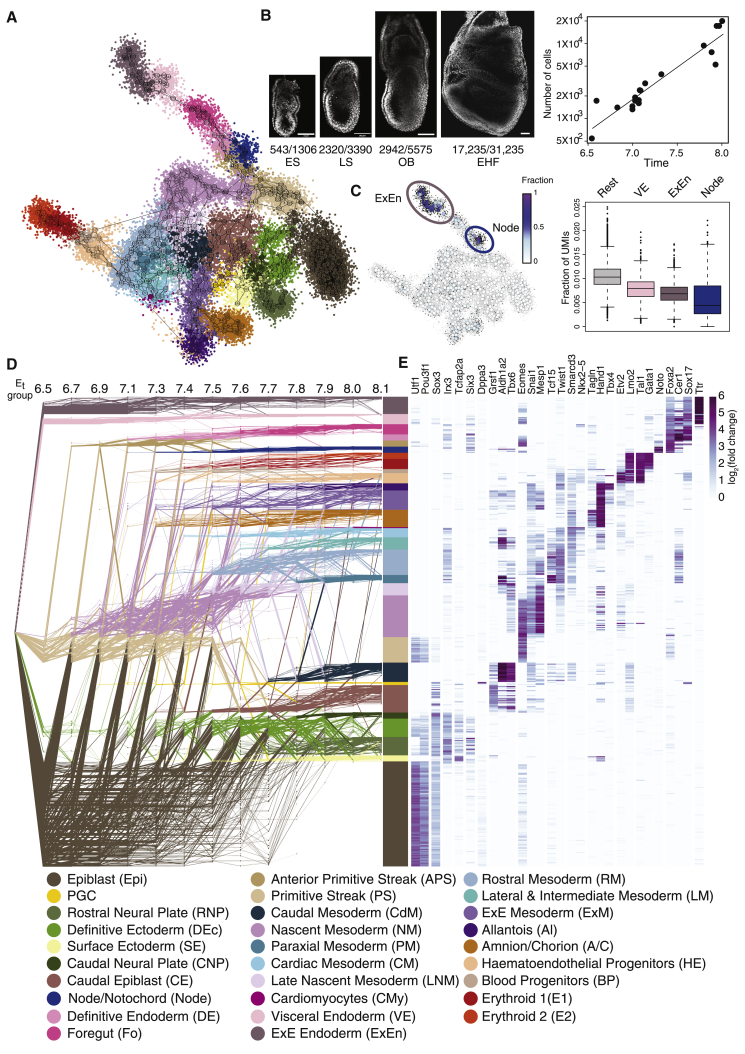
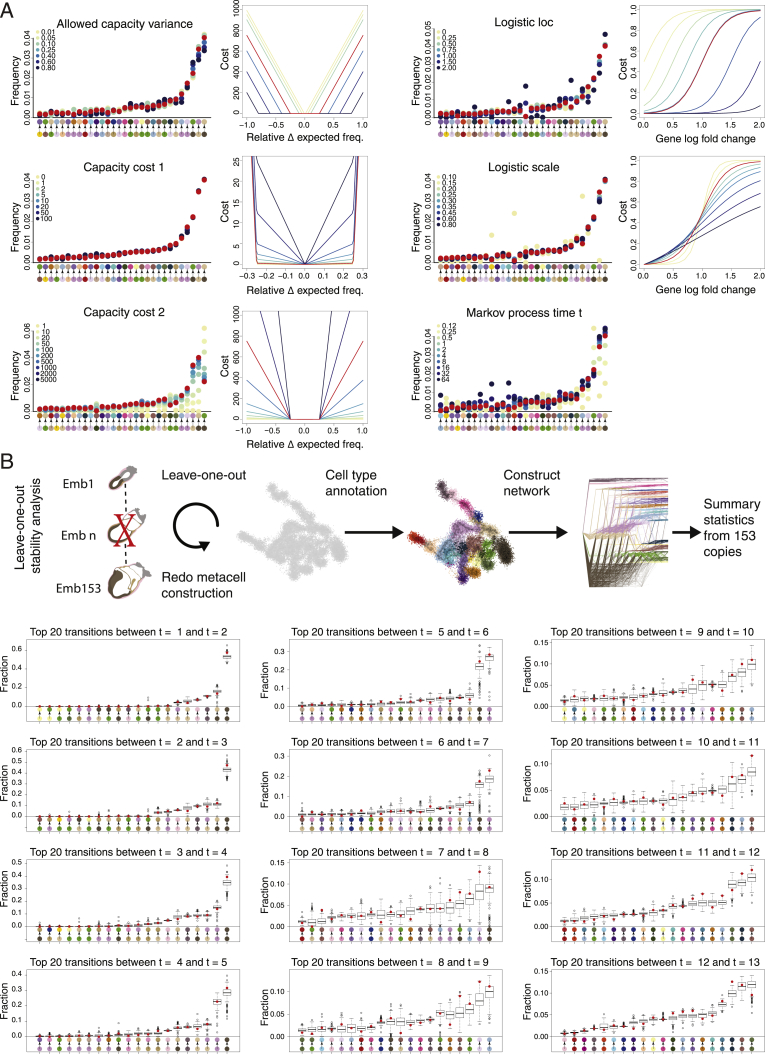
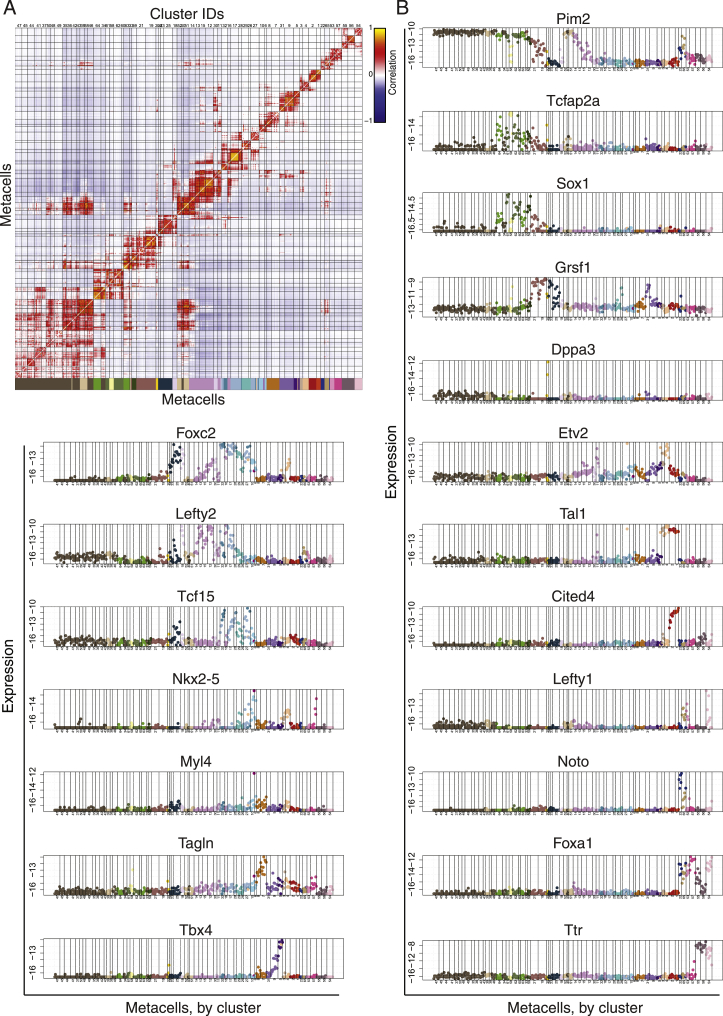
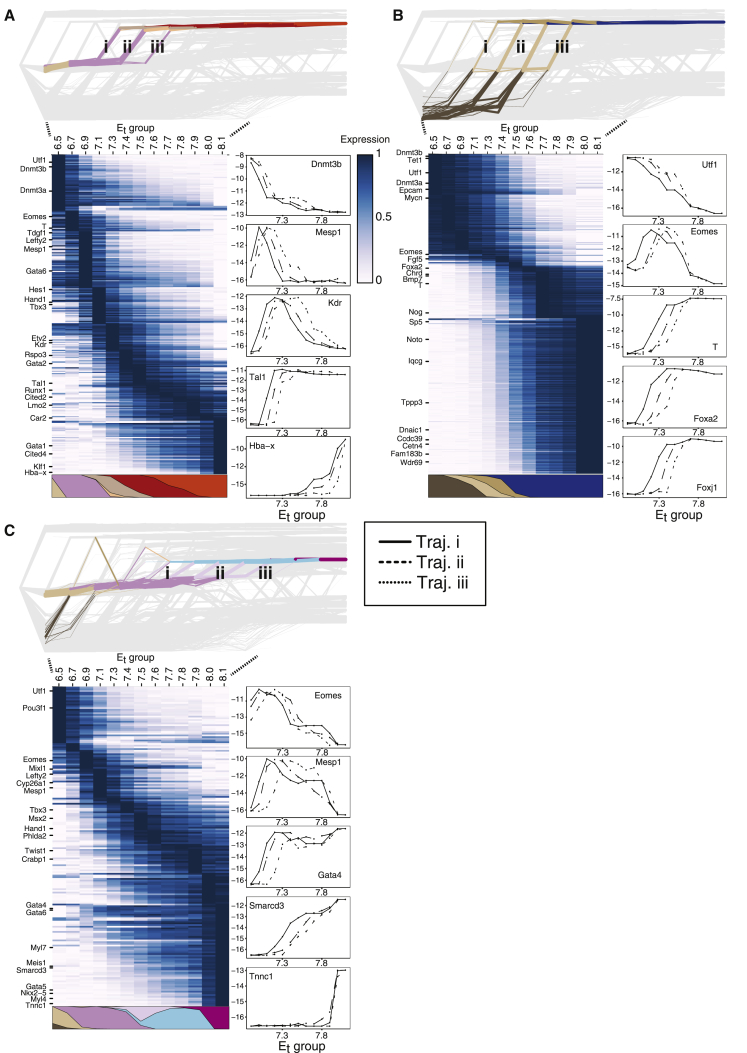
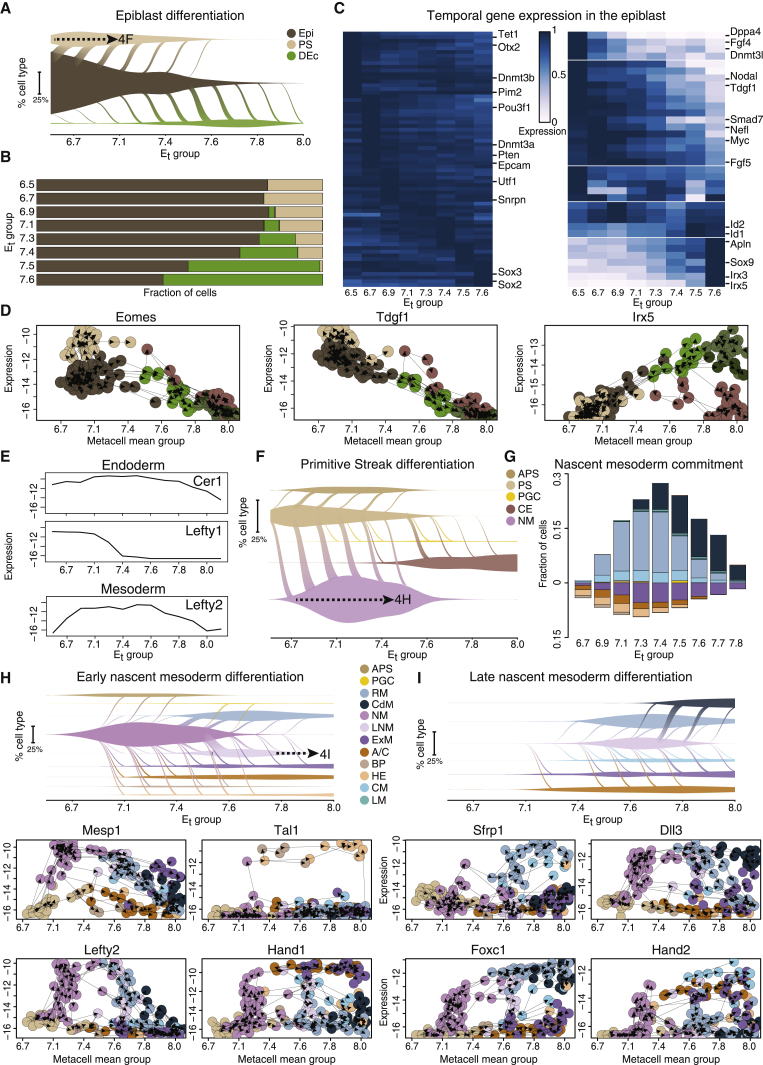
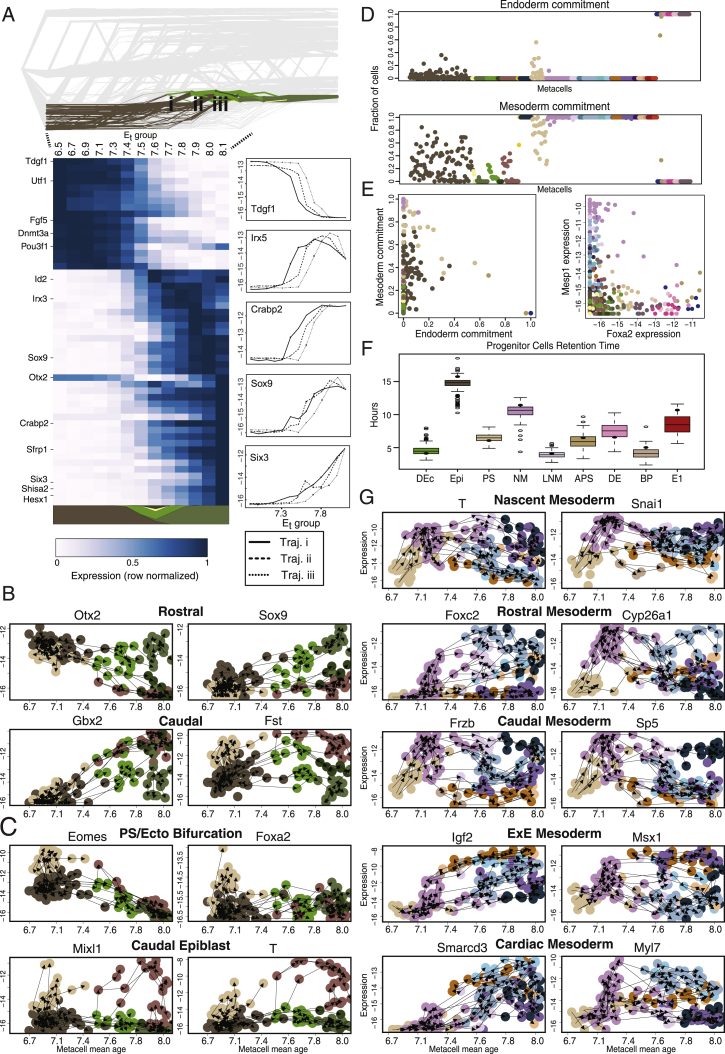
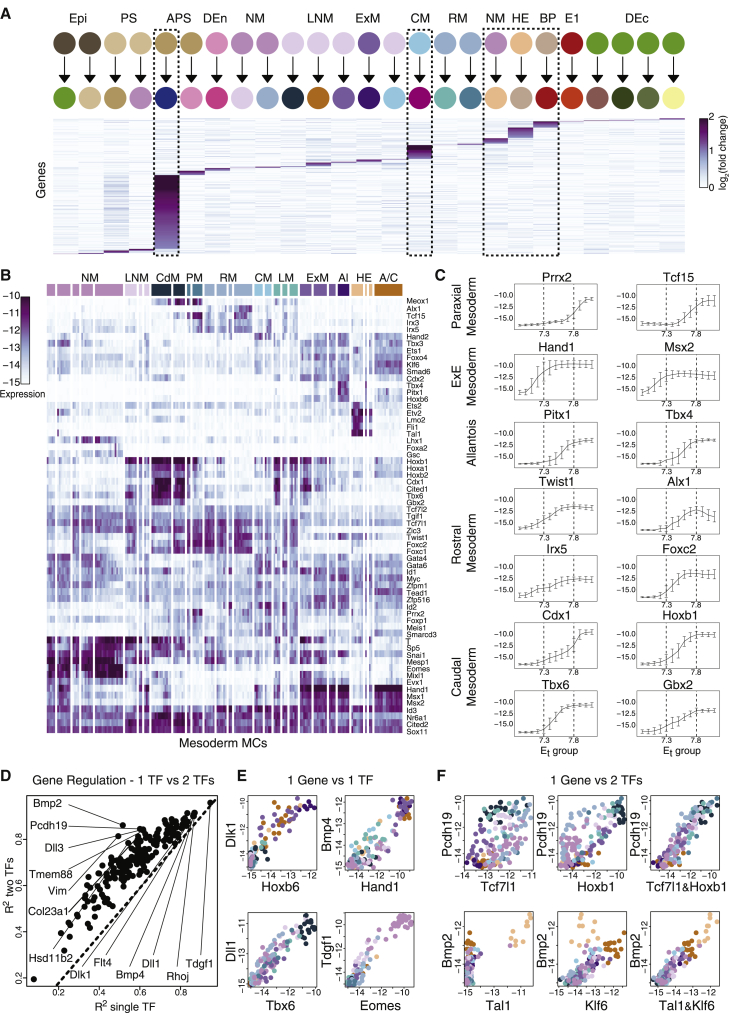
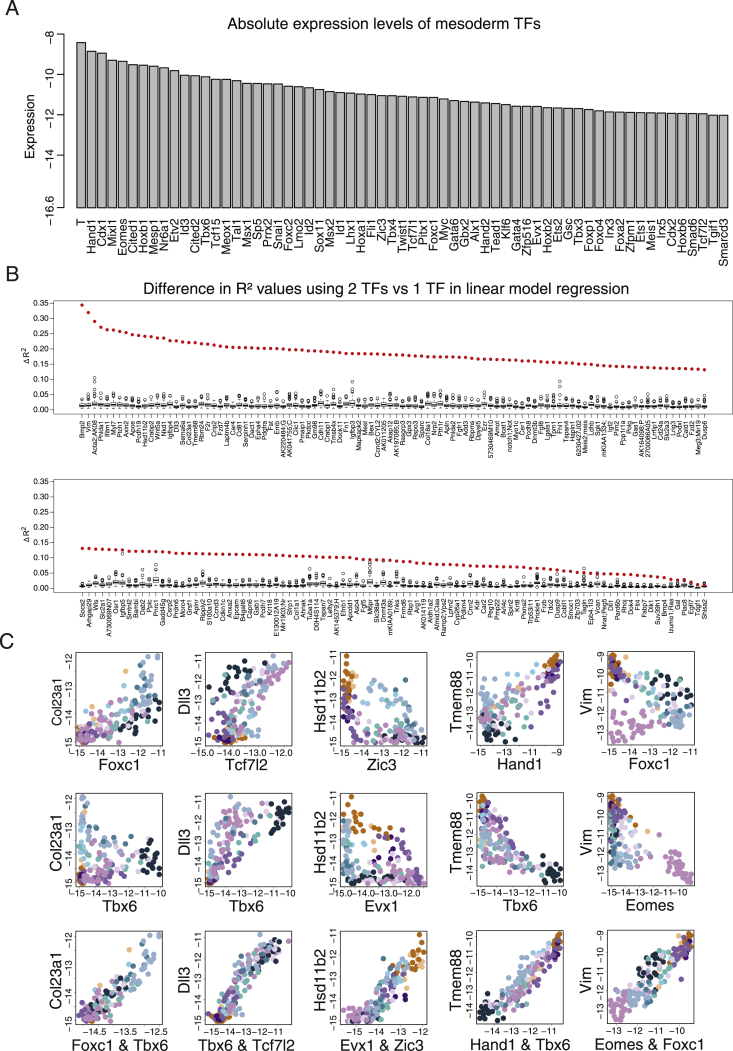
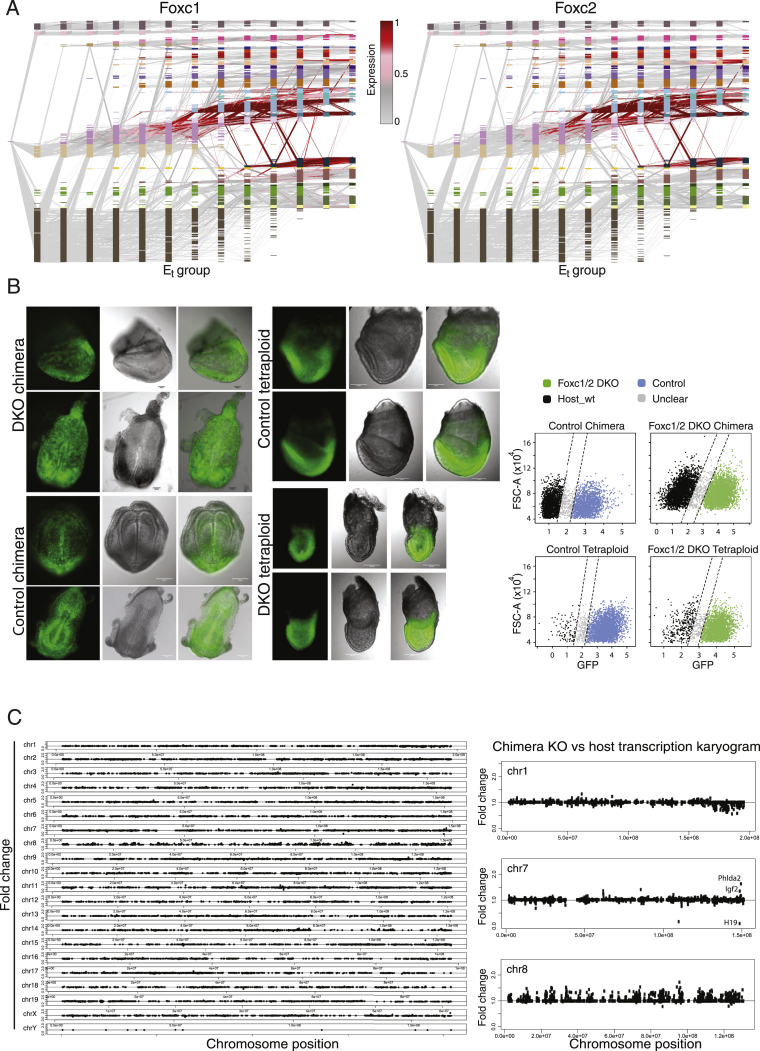
Mouse embryonic development is a canonical model system for studying mammalian cell fate acquisition. Recently, single-cell atlases comprehensively charted embryonic transcriptional landscapes, yet inference of the coordinated dynamics of cells over such atlases remains challenging. Here, we introduce a temporal model for mouse gastrulation, consisting of data from 153 individually sampled embryos spanning 36 h of molecular diversification. Using algorithms and precise timing, we infer differentiation flows and lineage specification dynamics over the embryonic transcriptional manifold. Rapid transcriptional bifurcations characterize the commitment of early specialized node and blood cells. However, for most lineages, we observe combinatorial multi-furcation dynamics rather than hierarchical transcriptional transitions. In the mesoderm, dozens of transcription factors combinatorially regulate multifurcations, as we exemplify using time-matched chimeric embryos of Foxc1/Foxc2 mutants. Our study rejects the notion of differentiation being governed by a series of binary choices, providing an alternative quantitative model for cell fate acquisition.
Keywords: cell fate decisions; chimera assay; developmental biology; mouse gastrulation; network flow model; scRNA-seq; tetraploid complementation assay; trajectory inference.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
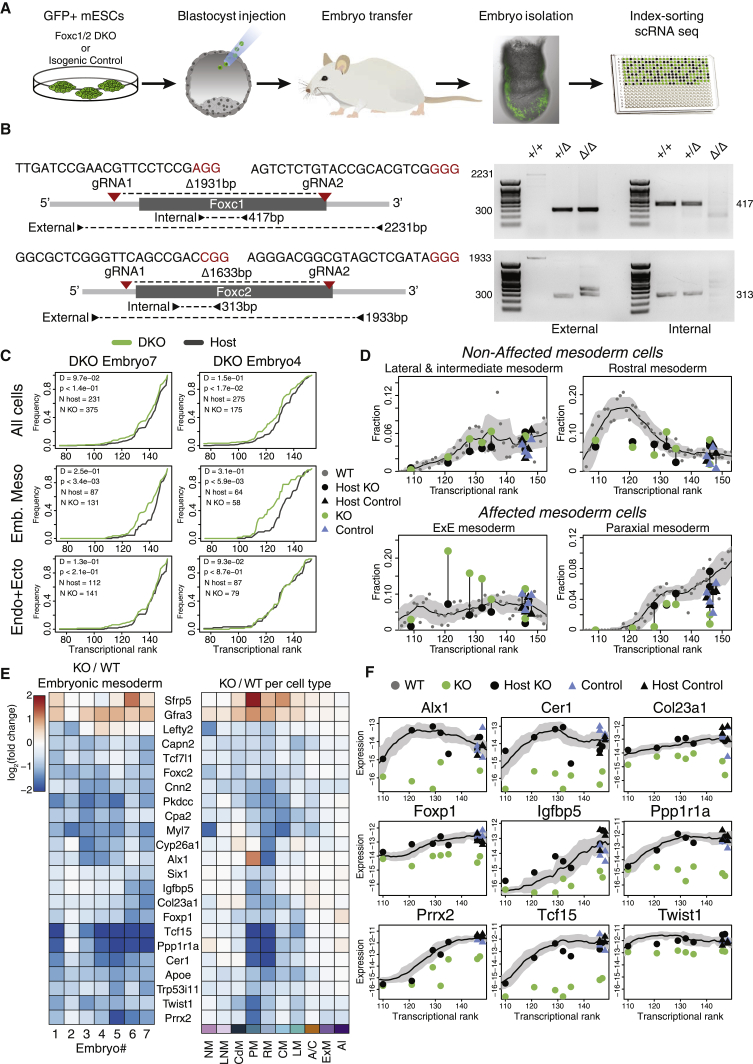
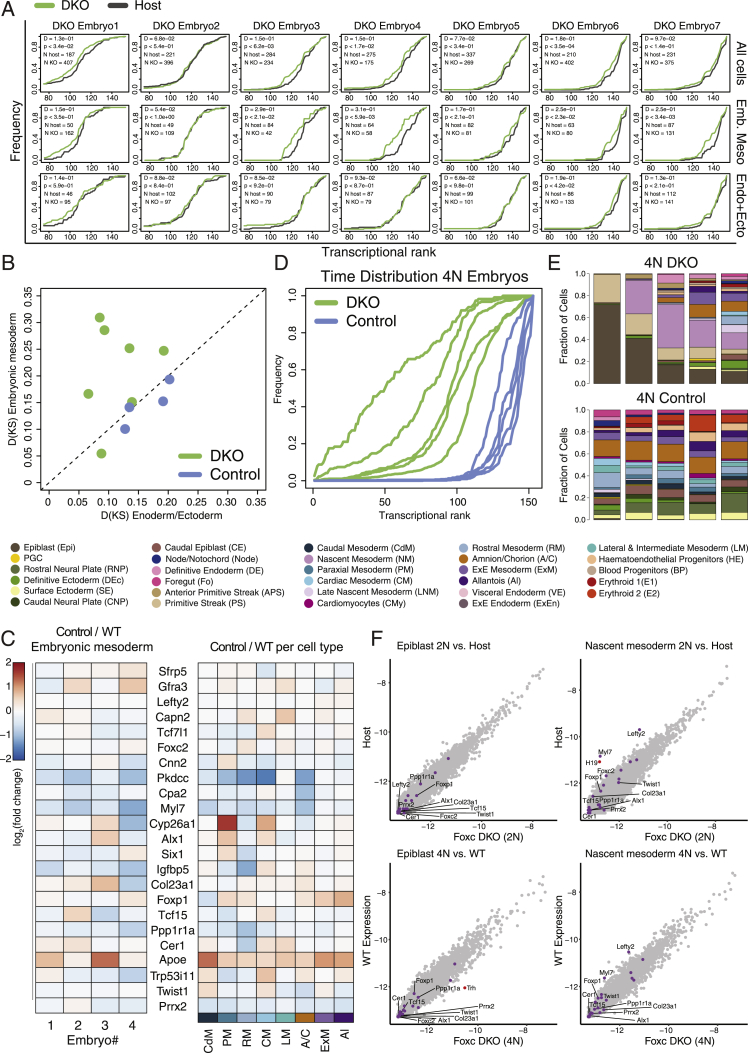
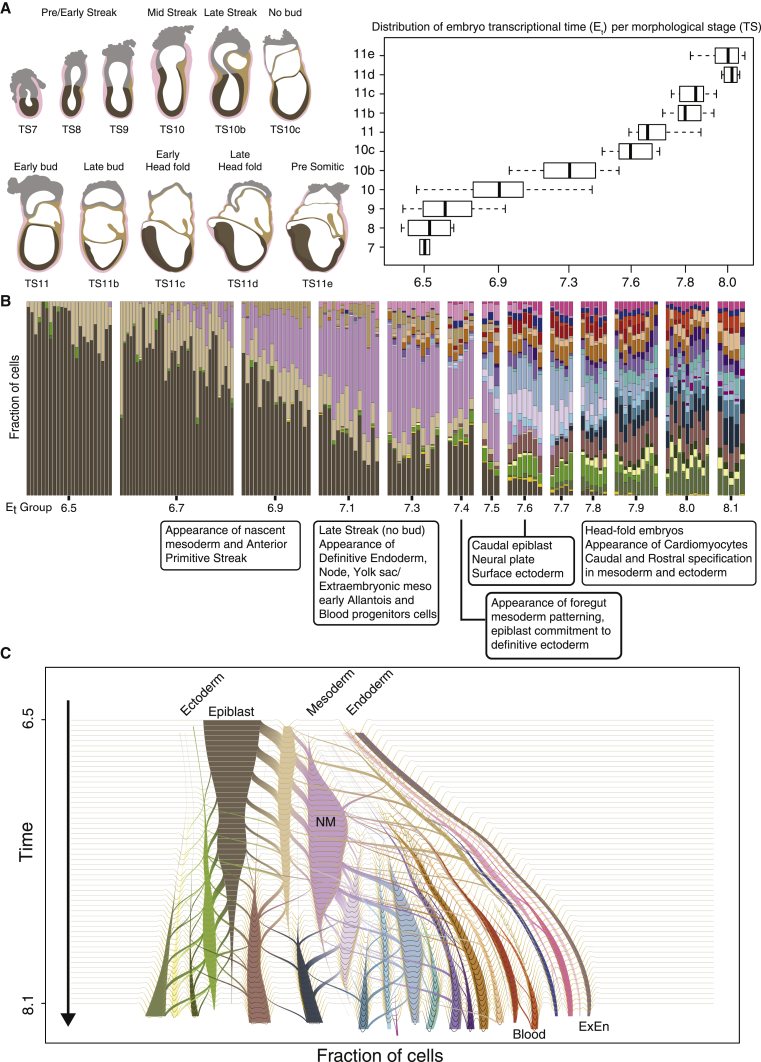
Comment in
-
A continuous model of early mammalian development.Nature. 2021 May;593(7858):200-201. doi: 10.1038/d41586-021-01153-1. Nature. 2021. PMID: 33931776 No abstract available.
-
Distributed and centralized control during differentiation.Dev Cell. 2021 Aug 9;56(15):2142-2144. doi: 10.1016/j.devcel.2021.07.007. Dev Cell. 2021. PMID: 34375580
References
-
- Ahuja R.K., Magnanti T.L., Orlin J.B. Prentice Hall; 1993. Network Flows: Theory, Algorithms and Applications.
-
- Arnold S.J., Robertson E.J. Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nat. Rev. Mol. Cell Biol. 2009;10:91–103. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
