

MRCKβ links Dasm1 to actin rearrangements to promote dendrite development
- PMID: 33933448
- PMCID: PMC8191314
- DOI: 10.1016/j.jbc.2021.100730
MRCKβ links Dasm1 to actin rearrangements to promote dendrite development
Abstract
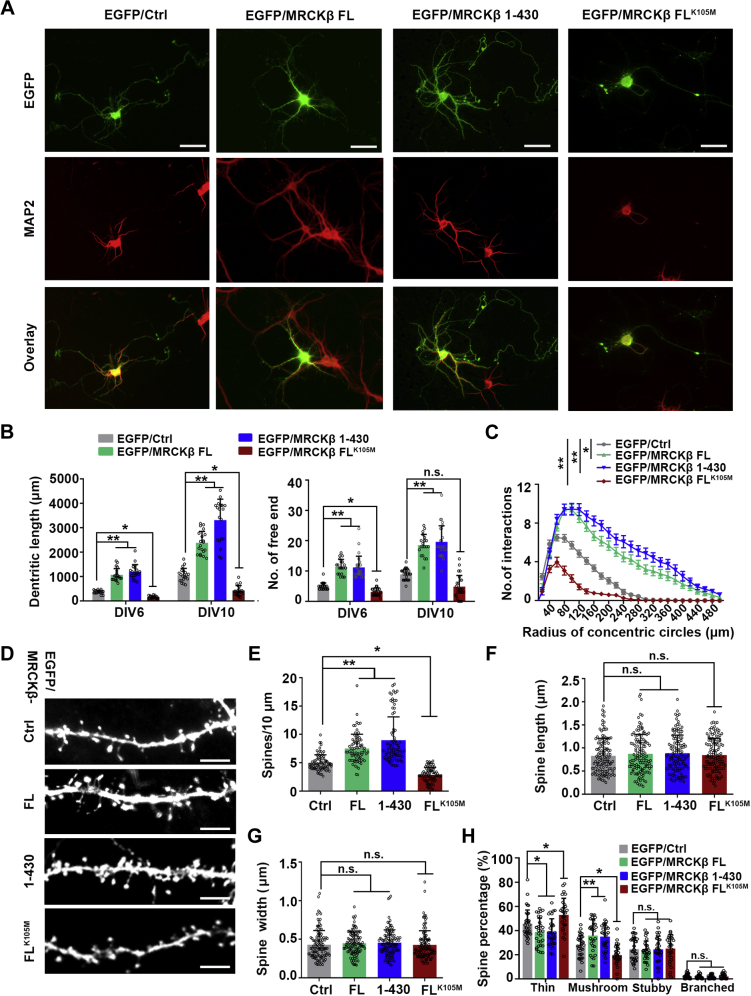
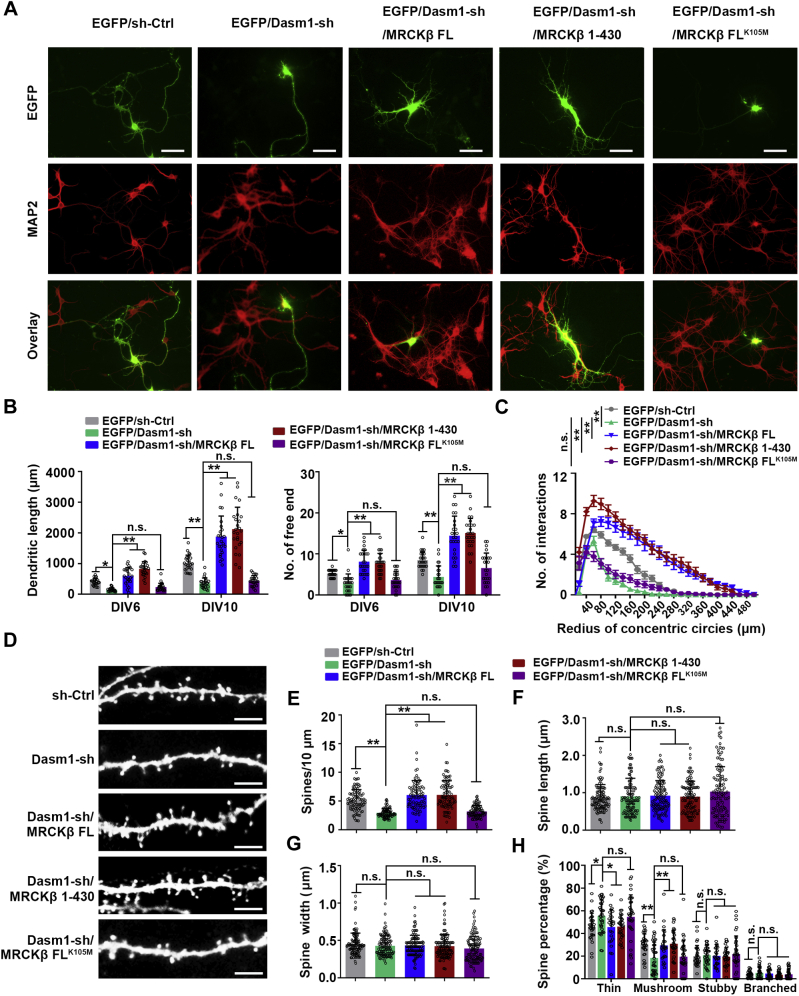
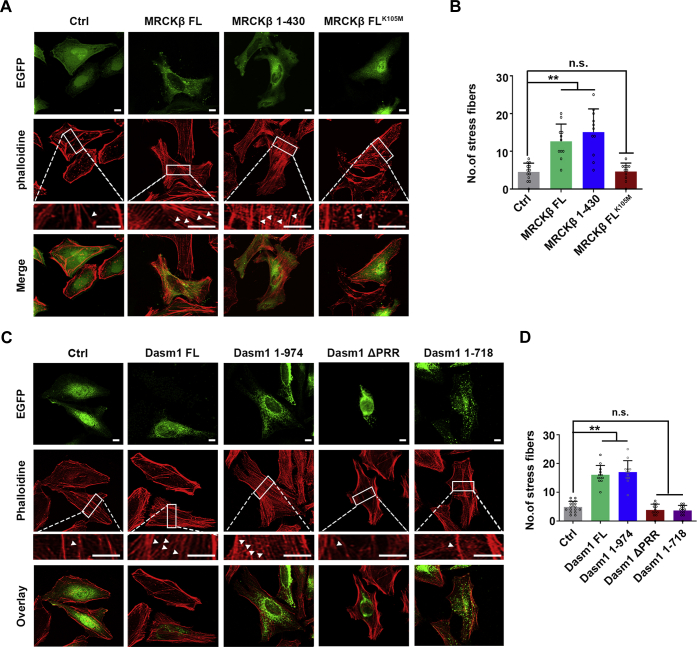
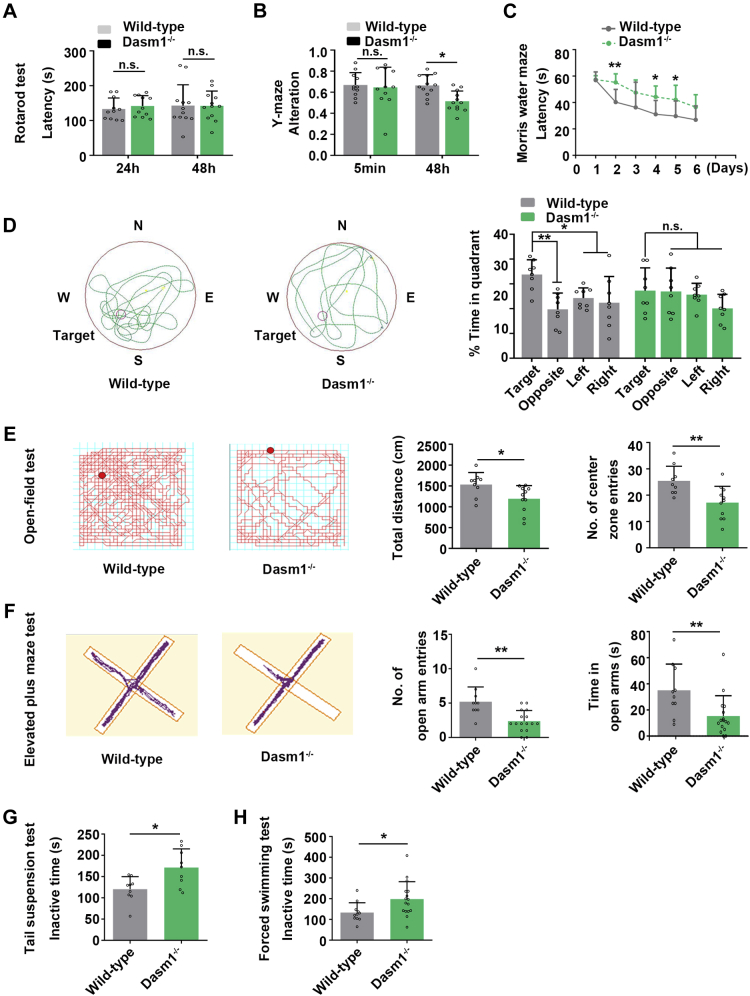
Proper dendrite morphogenesis and synapse formation are essential for neuronal development and function. Dasm1, a member of the immunoglobulin superfamily, is known to promote dendrite outgrowth and excitatory synapse maturation in vitro. However, the in vivo function of Dasm1 in neuronal development and the underlying mechanisms are not well understood. To learn more, Dasm1 knockout mice were constructed and employed to confirm that Dasm1 regulates dendrite arborization and spine formation in vivo. We performed a yeast two-hybrid screen using Dasm1, revealing MRCKβ as a putative partner; additional lines of evidence confirmed this interaction and identified cytoplasmic proline-rich region (823-947 aa) of Dasm1 and MRCKβ self-activated kinase domain (CC1, 410-744 aa) as necessary and sufficient for binding. Using co-immunoprecipitation assay, autophosphorylation assay, and BS3 cross-linking assay, we show that Dasm1 binding triggers a change in MRCKβ's conformation and subsequent dimerization, resulting in autophosphorylation and activation. Activated MRCKβ in turn phosphorylates a class 2 regulatory myosin light chain, which leads to enhanced actin rearrangement, causing the dendrite outgrowth and spine formation observed before. Removal of Dasm1 in mice leads to behavioral abnormalities. Together, these results reveal a crucial molecular pathway mediating cell surface and intracellular signaling communication to regulate actin dynamics and neuronal development in the mammalian brain.
Keywords: Dasm1; MRCKβ; actin rearrangement; dendrite development; spine formation.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures




References
-
- Magee J.C. Dendritic integration of excitatory synaptic input. Nat. Rev. Neurosci. 2000;1:181–190. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
