

The mammalian cholesterol synthesis enzyme squalene monooxygenase is proteasomally truncated to a constitutively active form
- PMID: 33933449
- PMCID: PMC8166775
- DOI: 10.1016/j.jbc.2021.100731
The mammalian cholesterol synthesis enzyme squalene monooxygenase is proteasomally truncated to a constitutively active form
Abstract
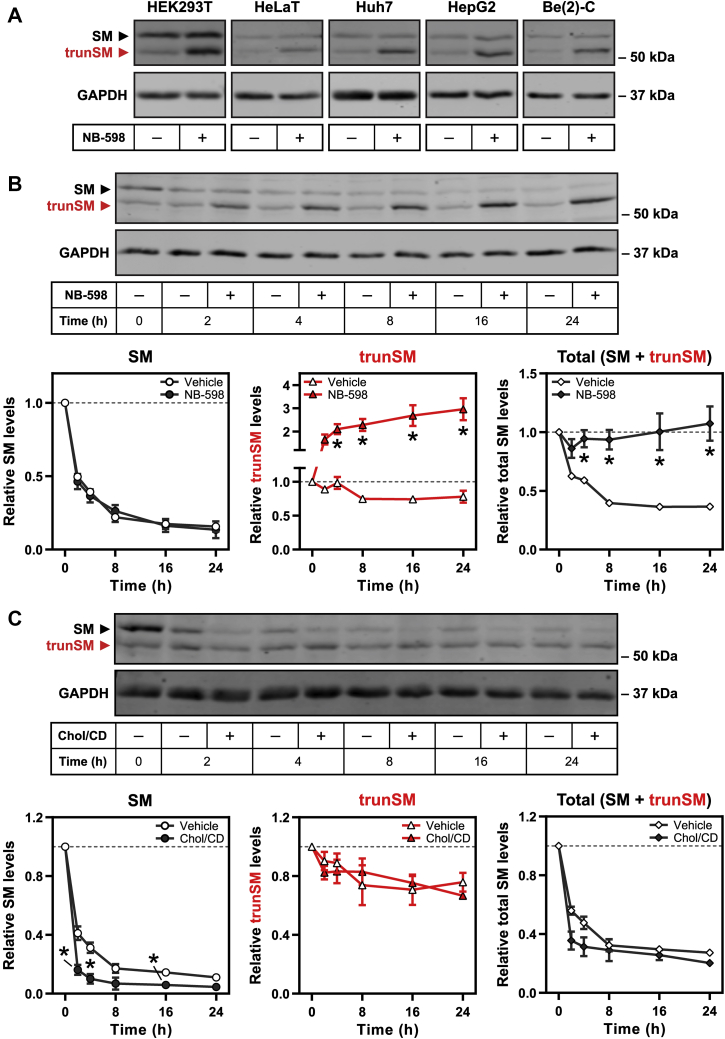
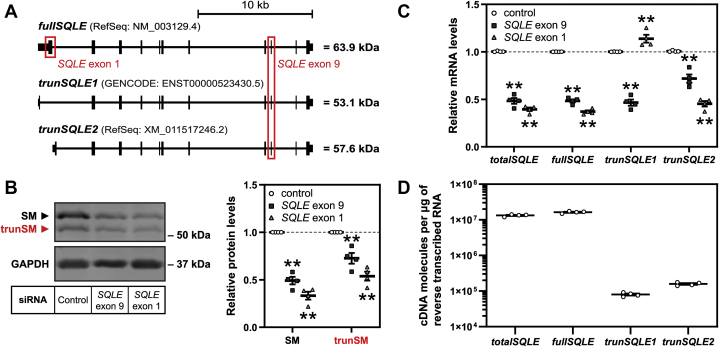
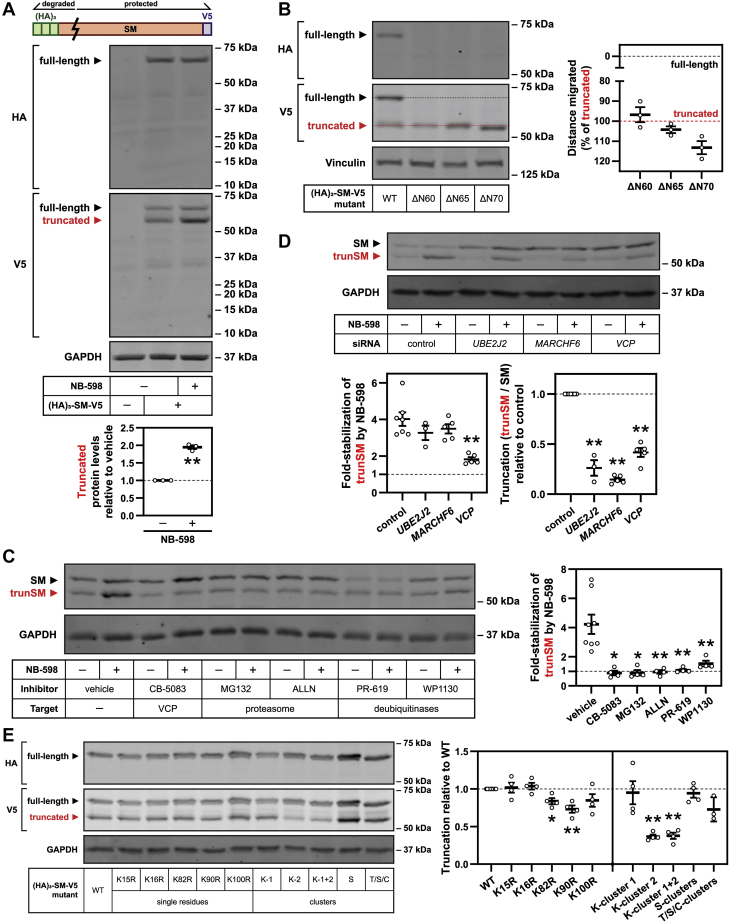
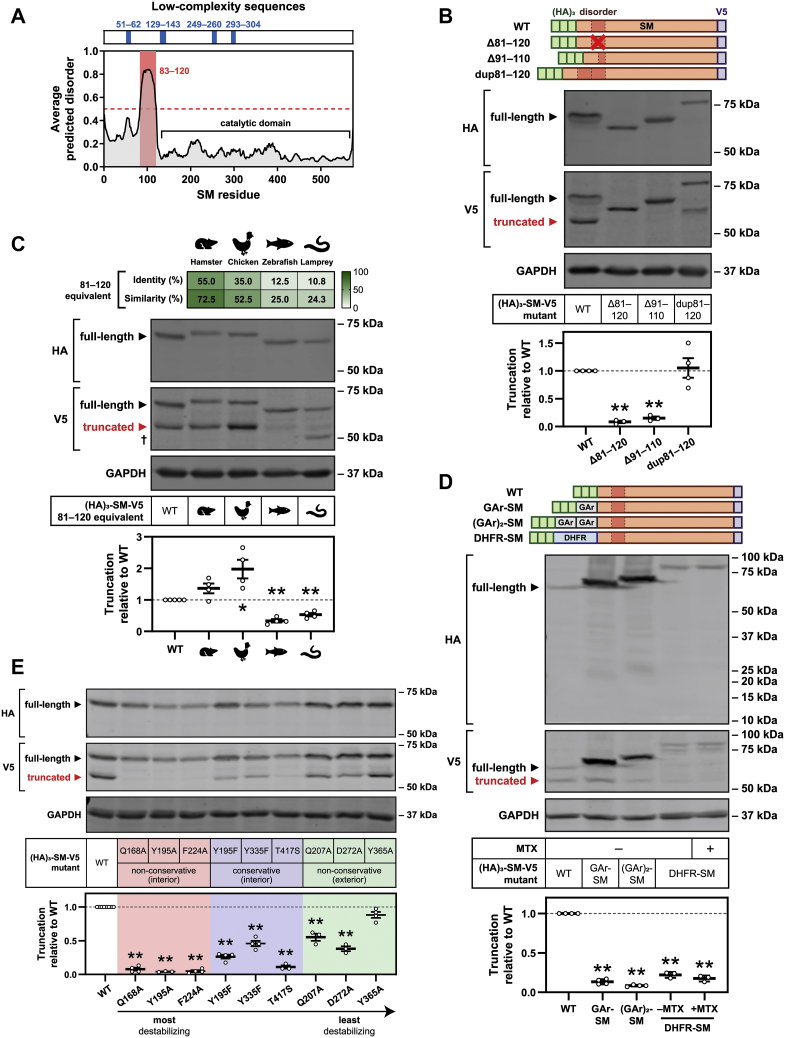
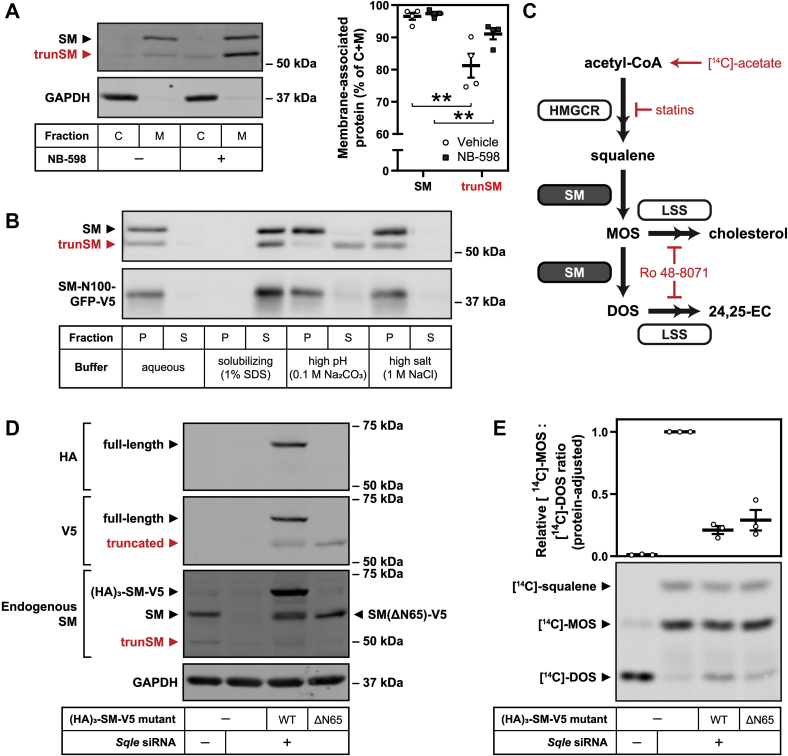
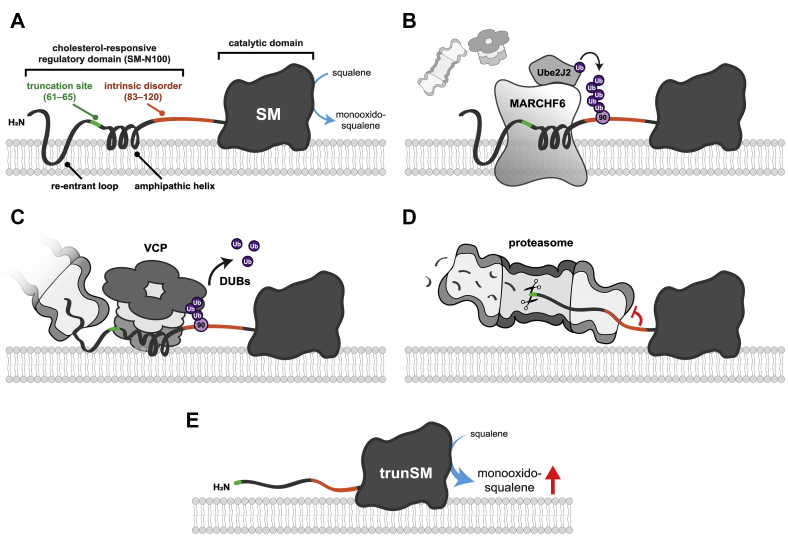
Squalene monooxygenase (SM, also known as squalene epoxidase) is a rate-limiting enzyme of cholesterol synthesis that converts squalene to monooxidosqualene and is oncogenic in numerous cancer types. SM is subject to feedback regulation via cholesterol-induced proteasomal degradation, which depends on its lipid-sensing N-terminal regulatory domain. We previously identified an endogenous truncated form of SM with a similar abundance to full-length SM, but whether this truncated form is functional or subject to the same regulatory mechanisms as full-length SM is not known. Here, we show that truncated SM differs from full-length SM in two major ways: it is cholesterol resistant and adopts a peripheral rather than integral association with the endoplasmic reticulum membrane. However, truncated SM retains full SM activity and is therefore constitutively active. Truncation of SM occurs during its endoplasmic reticulum-associated degradation and requires the proteasome, which partially degrades the SM N-terminus and disrupts cholesterol-sensing elements within the regulatory domain. Furthermore, truncation relies on a ubiquitin signal that is distinct from that required for cholesterol-induced degradation. Using mutagenesis, we demonstrate that partial proteasomal degradation of SM depends on both an intrinsically disordered region near the truncation site and the stability of the adjacent catalytic domain, which escapes degradation. These findings uncover an additional layer of complexity in the post-translational regulation of cholesterol synthesis and establish SM as the first eukaryotic enzyme found to undergo proteasomal truncation.
Keywords: cholesterol; endoplasmic reticulum–associated protein degradation; proteasome; protein degradation; squalene monooxygenase; ubiquitylation (ubiquitination).
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008;9:125–138. - PubMed
-
- Prospective Studies Collaboration Blood cholesterol and vascular mortality by age, sex, and blood pressure: A meta-analysis of individual data from 61 prospective studies with 55 000 vascular deaths. Lancet. 2007;370:1829–1839. - PubMed
-
- Howe V., Sharpe L.J., Alexopoulos S.J., Kunze S.V., Chua N.K., Li D., Brown A.J. Cholesterol homeostasis: How do cells sense sterol excess? Chem. Phys. Lipids. 2016;199:170–178. - PubMed
-
- Gill S., Stevenson J., Kristiana I., Brown A.J. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 2011;13:260–273. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
