

Human methylome variation across Infinium 450K data on the Gene Expression Omnibus
- PMID: 33937763
- PMCID: PMC8061458
- DOI: 10.1093/nargab/lqab025
Human methylome variation across Infinium 450K data on the Gene Expression Omnibus
Abstract
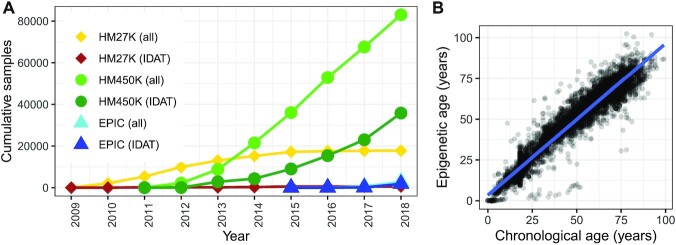
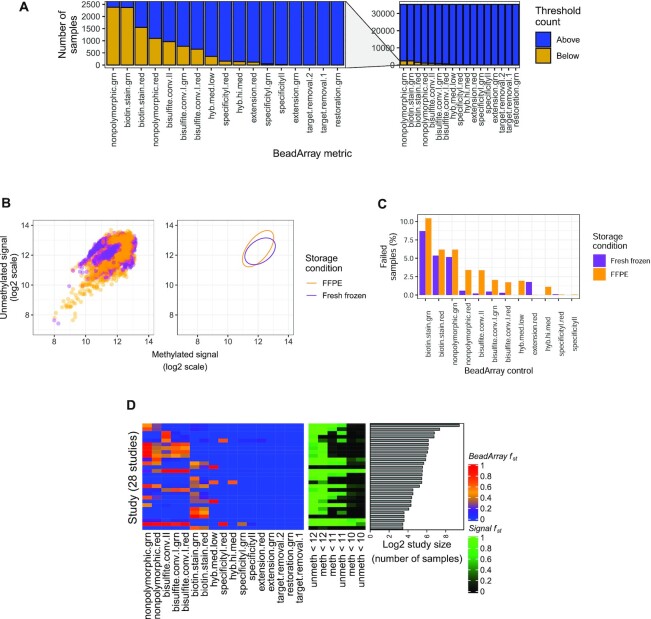
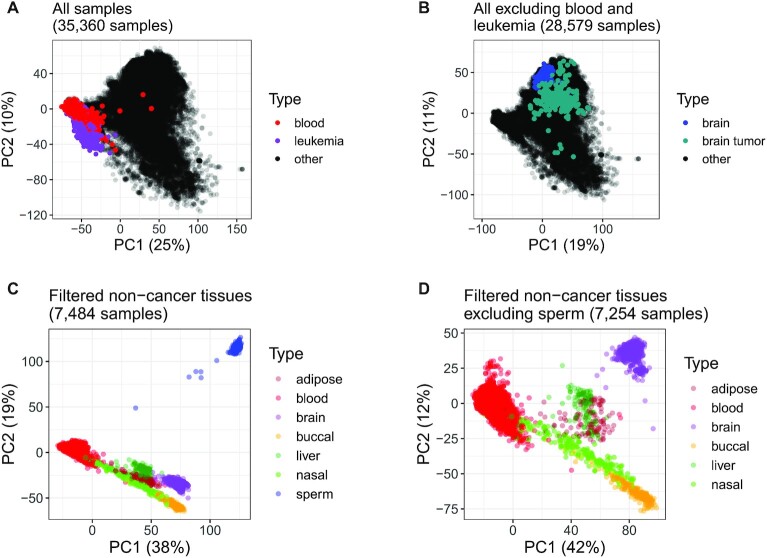
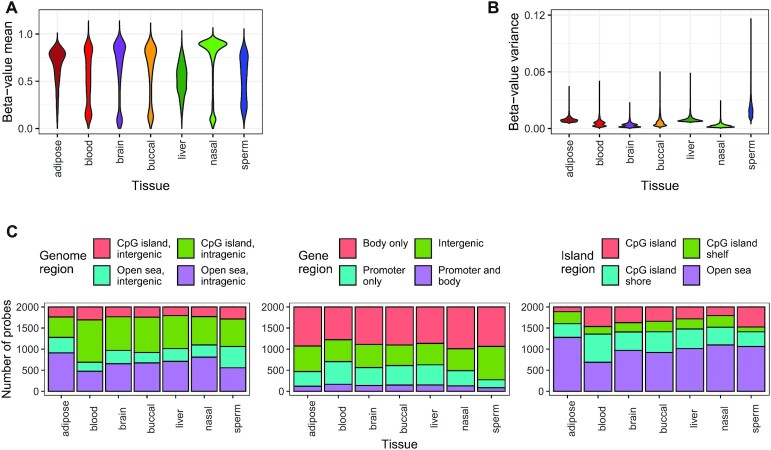
While DNA methylation (DNAm) is the most-studied epigenetic mark, few recent studies probe the breadth of publicly available DNAm array samples. We collectively analyzed 35 360 Illumina Infinium HumanMethylation450K DNAm array samples published on the Gene Expression Omnibus. We learned a controlled vocabulary of sample labels by applying regular expressions to metadata and used existing models to predict various sample properties including epigenetic age. We found approximately two-thirds of samples were from blood, one-quarter were from brain and one-third were from cancer patients. About 19% of samples failed at least one of Illumina's 17 prescribed quality assessments; signal distributions across samples suggest modifying manufacturer-recommended thresholds for failure would make these assessments more informative. We further analyzed DNAm variances in seven tissues (adipose, nasal, blood, brain, buccal, sperm and liver) and characterized specific probes distinguishing them. Finally, we compiled DNAm array data and metadata, including our learned and predicted sample labels, into database files accessible via the recountmethylation R/Bioconductor companion package. Its vignettes walk the user through some analyses contained in this paper.
© The Author(s) 2021. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
