

Functional and genetic determinants of mutation rate variability in regulatory elements of cancer genomes
- PMID: 33941236
- PMCID: PMC8091793
- DOI: 10.1186/s13059-021-02318-x
Functional and genetic determinants of mutation rate variability in regulatory elements of cancer genomes
Abstract
Background: Cancer genomes are shaped by mutational processes with complex spatial variation at multiple scales. Entire classes of regulatory elements are affected by local variations in mutation frequency. However, the underlying mechanisms with functional and genetic determinants remain poorly understood.
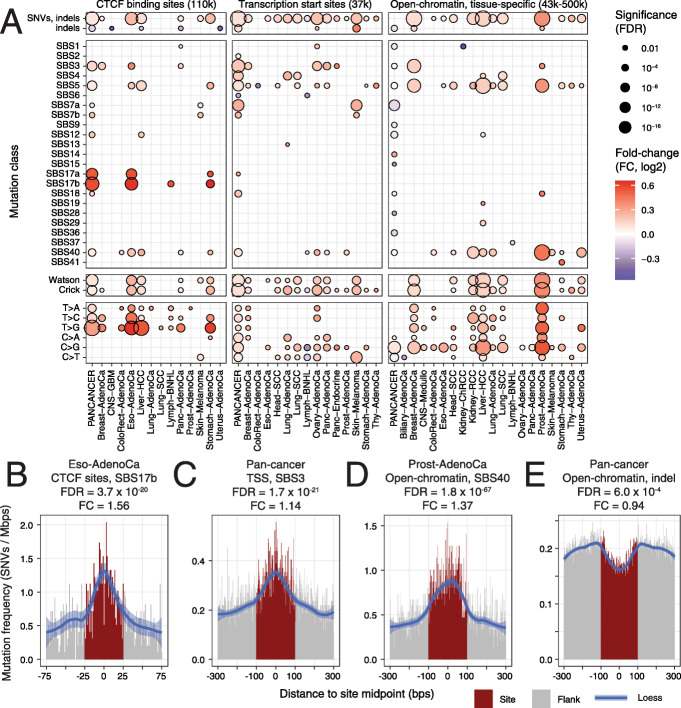
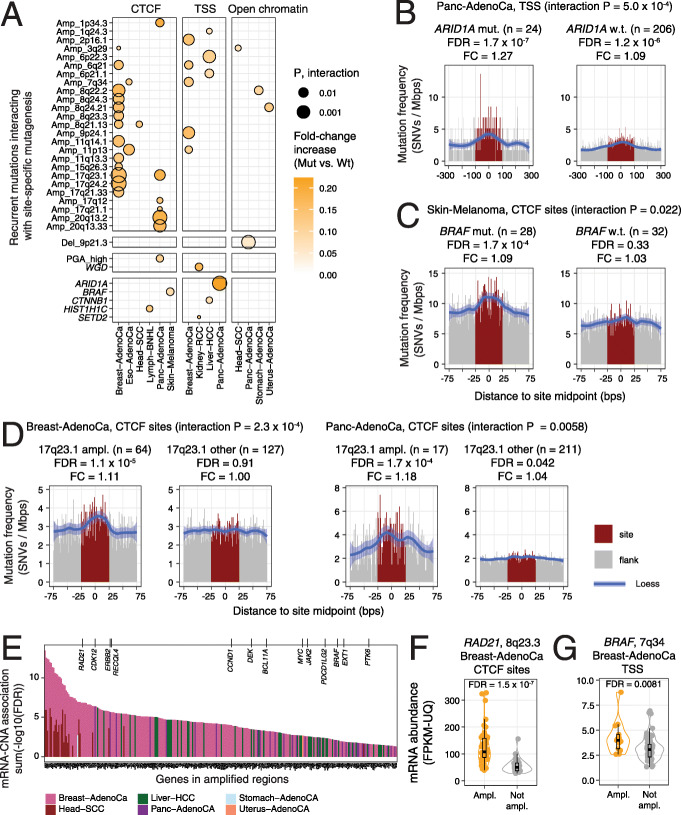
Results: We characterise the mutational landscape of 1.3 million gene-regulatory and chromatin architectural elements in 2419 whole cancer genomes with transcriptional and pathway activity, functional conservation and recurrent driver events. We develop RM2, a statistical model that quantifies mutational enrichment or depletion in classes of genomic elements through genetic, trinucleotide and megabase-scale effects. We report a map of localised mutational processes affecting CTCF binding sites, transcription start sites (TSS) and tissue-specific open-chromatin regions. Increased mutation frequency in TSSs associates with mRNA abundance in most cancer types, while open-chromatin regions are generally enriched in mutations. We identify ~ 10,000 CTCF binding sites with core DNA motifs and constitutive binding in 66 cell types that represent focal points of mutagenesis. We detect site-specific mutational signature enrichments, such as SBS40 in open-chromatin regions in prostate cancer and SBS17b in CTCF binding sites in gastrointestinal cancers. Candidate drivers of localised mutagenesis are also apparent: BRAF mutations associate with mutational enrichments at CTCF binding sites in melanoma, and ARID1A mutations with TSS-specific mutagenesis in pancreatic cancer.
Conclusions: Our method and catalogue of localised mutational processes provide novel perspectives to cancer genome evolution, mutagenesis, DNA repair and driver gene discovery. The functional and genetic correlates of mutational processes suggest mechanistic hypotheses for future studies.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
