

Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype
- PMID: 33941880
- PMCID: PMC8354849
- DOI: 10.1038/s41436-021-01158-1
Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype
Abstract
Purpose: Despite a few recent reports of patients harboring truncating variants in NSD2, a gene considered critical for the Wolf-Hirschhorn syndrome (WHS) phenotype, the clinical spectrum associated with NSD2 pathogenic variants remains poorly understood.
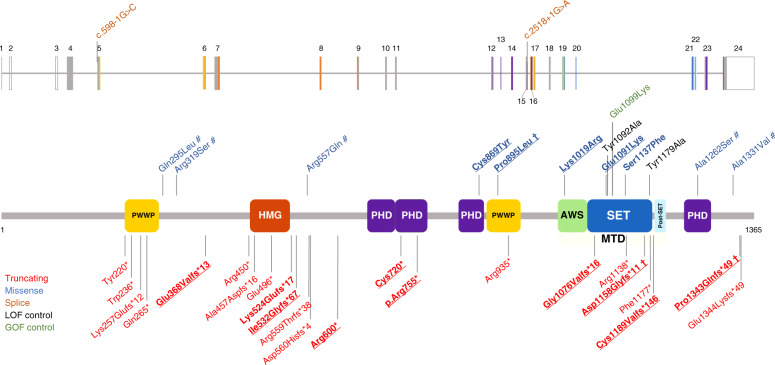
Methods: We collected a comprehensive series of 18 unpublished patients carrying heterozygous missense, elongating, or truncating NSD2 variants; compared their clinical data to the typical WHS phenotype after pooling them with ten previously described patients; and assessed the underlying molecular mechanism by structural modeling and measuring methylation activity in vitro.
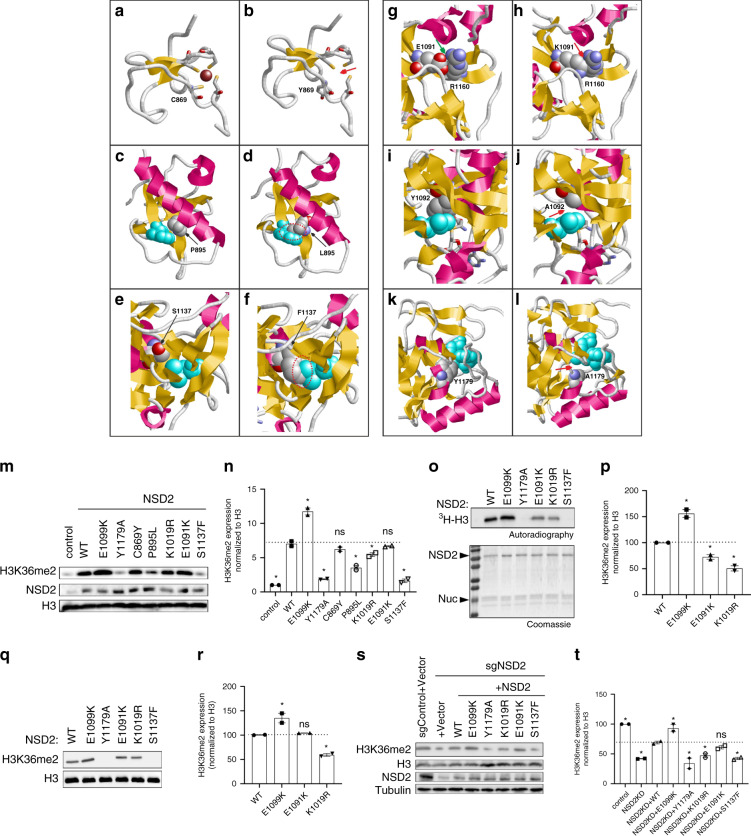
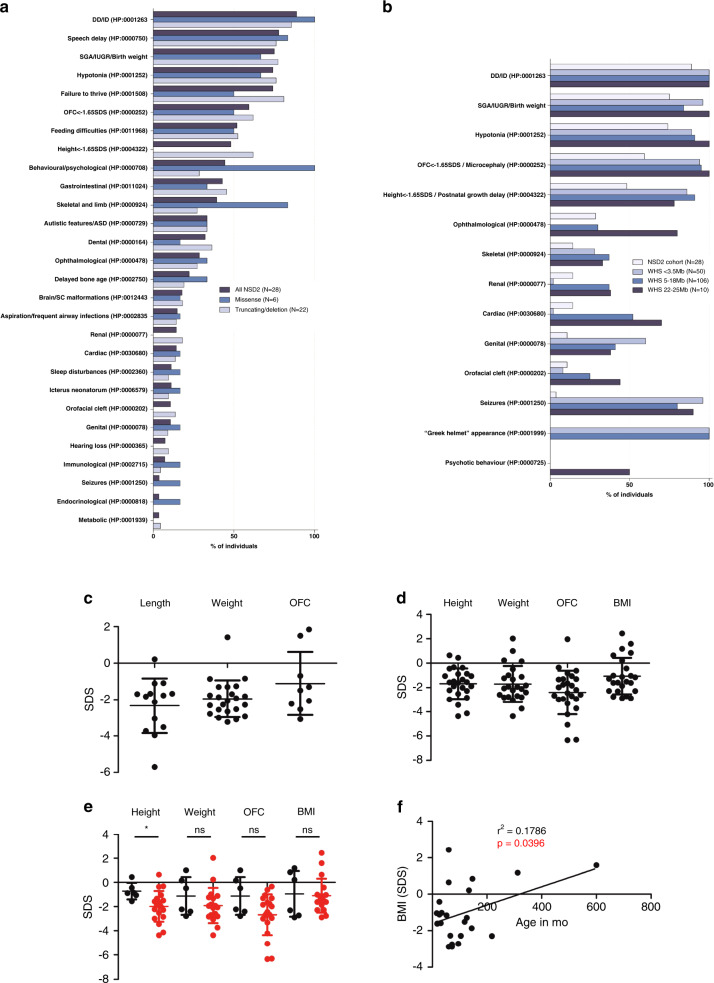
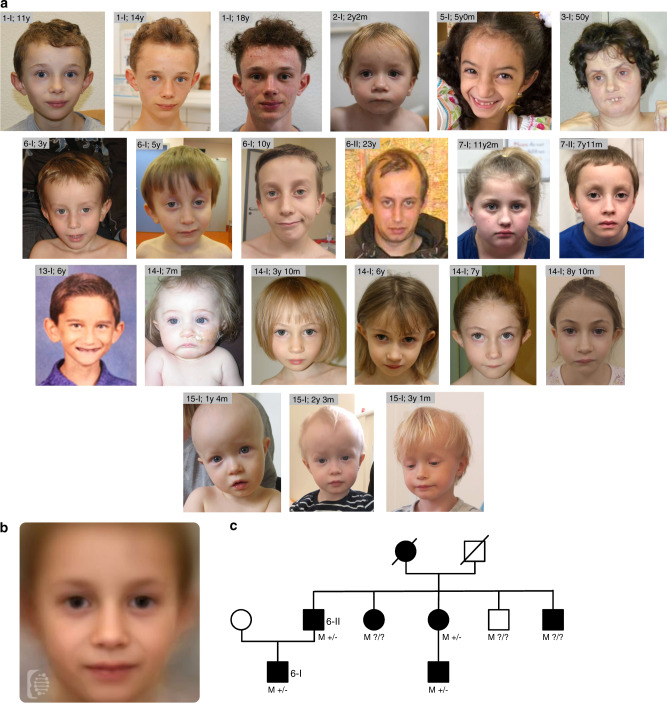
Results: The core NSD2-associated phenotype includes mostly mild developmental delay, prenatal-onset growth retardation, low body mass index, and characteristic facial features distinct from WHS. Patients carrying missense variants were significantly taller and had more frequent behavioral/psychological issues compared with those harboring truncating variants. Structural in silico modeling suggested interference with NSD2's folding and function for all missense variants in known structures. In vitro testing showed reduced methylation activity and failure to reconstitute H3K36me2 in NSD2 knockout cells for most missense variants.
Conclusion: NSD2 loss-of-function variants lead to a distinct, rather mild phenotype partially overlapping with WHS. To avoid confusion for patients, NSD2 deficiency may be named Rauch-Steindl syndrome after the delineators of this phenotype.
© 2021. The Author(s).
Conflict of interest statement
O.G. is a co-founder and member of the Board of Directors of EpiCypher, Inc. The other authors declare no competing interests.
Figures
Comment in
-
Correspondence on "Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype" by Zanoni et al.Genet Med. 2022 Mar;24(3):754-756. doi: 10.1016/j.gim.2021.11.007. Epub 2021 Dec 6. Genet Med. 2022. PMID: 34906509 No abstract available.
-
Response to Cueto-González et al.Genet Med. 2022 Mar;24(3):757. doi: 10.1016/j.gim.2021.11.006. Epub 2021 Dec 7. Genet Med. 2022. PMID: 34906511 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
