

An Evolutionary Portrait of the Progenitor SARS-CoV-2 and Its Dominant Offshoots in COVID-19 Pandemic
- PMID: 33942847
- PMCID: PMC8135569
- DOI: 10.1093/molbev/msab118
An Evolutionary Portrait of the Progenitor SARS-CoV-2 and Its Dominant Offshoots in COVID-19 Pandemic
Abstract
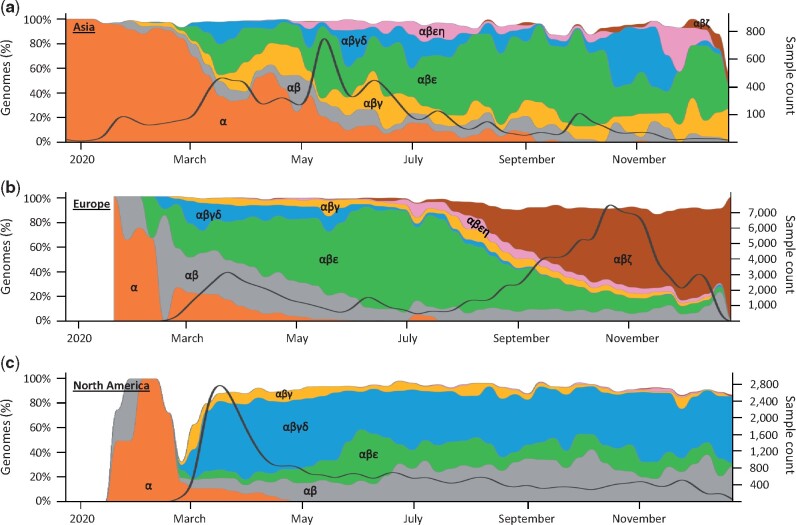
Global sequencing of genomes of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has continued to reveal new genetic variants that are the key to unraveling its early evolutionary history and tracking its global spread over time. Here we present the heretofore cryptic mutational history and spatiotemporal dynamics of SARS-CoV-2 from an analysis of thousands of high-quality genomes. We report the likely most recent common ancestor of SARS-CoV-2, reconstructed through a novel application and advancement of computational methods initially developed to infer the mutational history of tumor cells in a patient. This progenitor genome differs from genomes of the first coronaviruses sampled in China by three variants, implying that none of the earliest patients represent the index case or gave rise to all the human infections. However, multiple coronavirus infections in China and the United States harbored the progenitor genetic fingerprint in January 2020 and later, suggesting that the progenitor was spreading worldwide months before and after the first reported cases of COVID-19 in China. Mutations of the progenitor and its offshoots have produced many dominant coronavirus strains that have spread episodically over time. Fingerprinting based on common mutations reveals that the same coronavirus lineage has dominated North America for most of the pandemic in 2020. There have been multiple replacements of predominant coronavirus strains in Europe and Asia as well as continued presence of multiple high-frequency strains in Asia and North America. We have developed a continually updating dashboard of global evolution and spatiotemporal trends of SARS-CoV-2 spread (http://sars2evo.datamonkey.org/).
Keywords: coronavirus; phylogeny; web tool.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
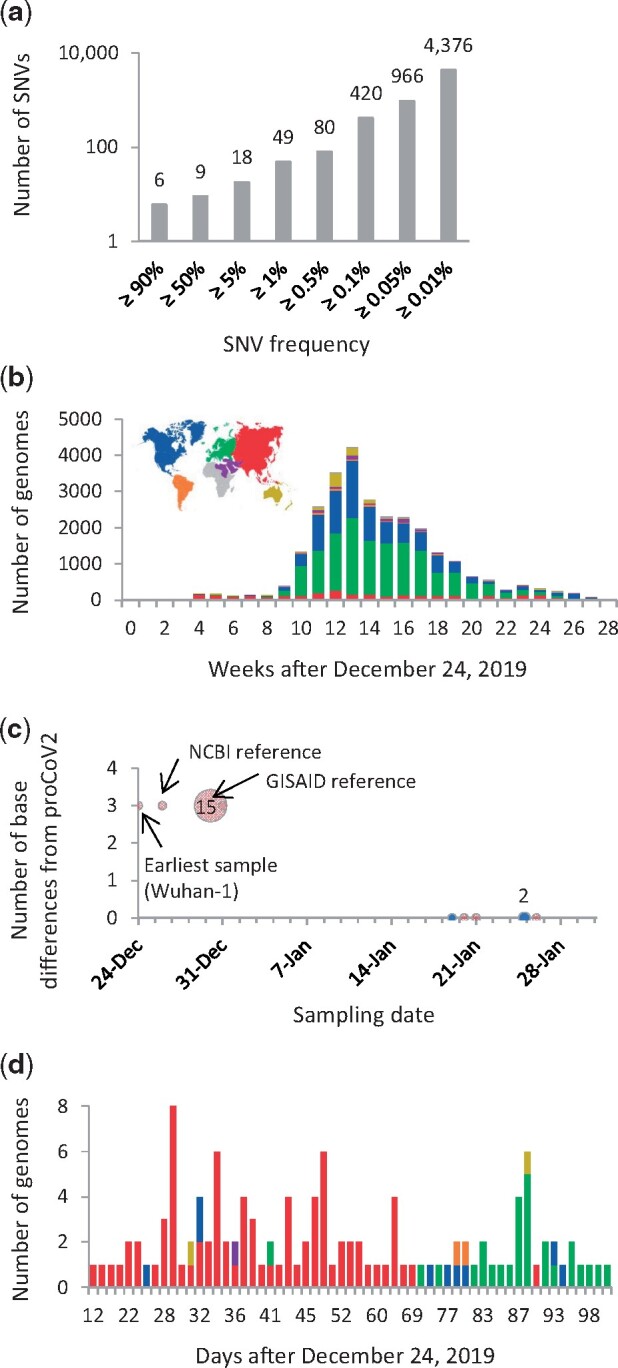
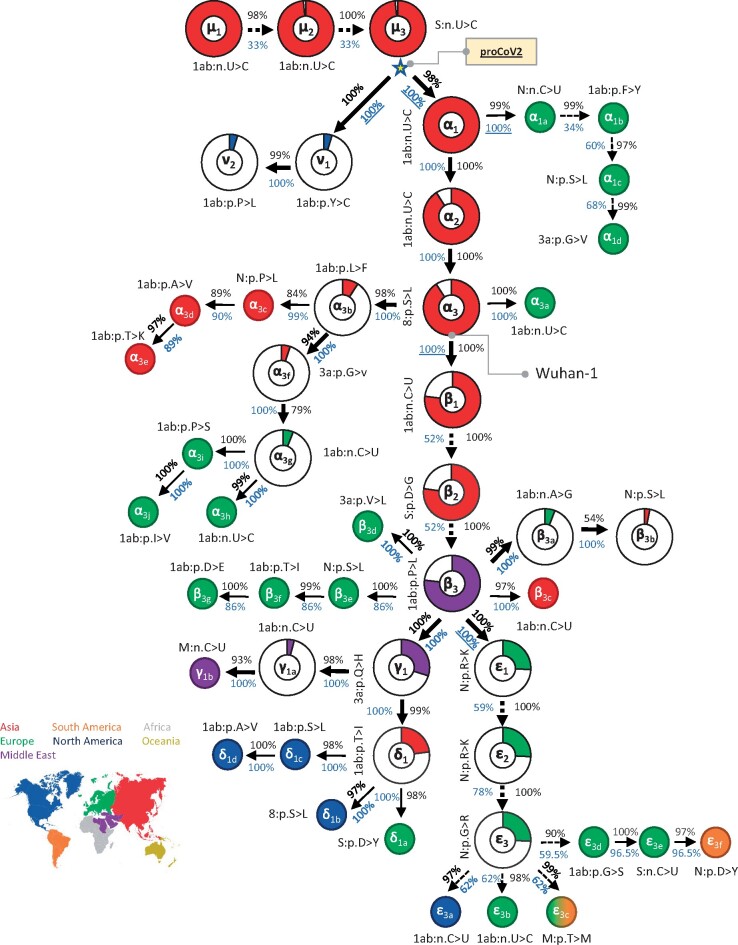
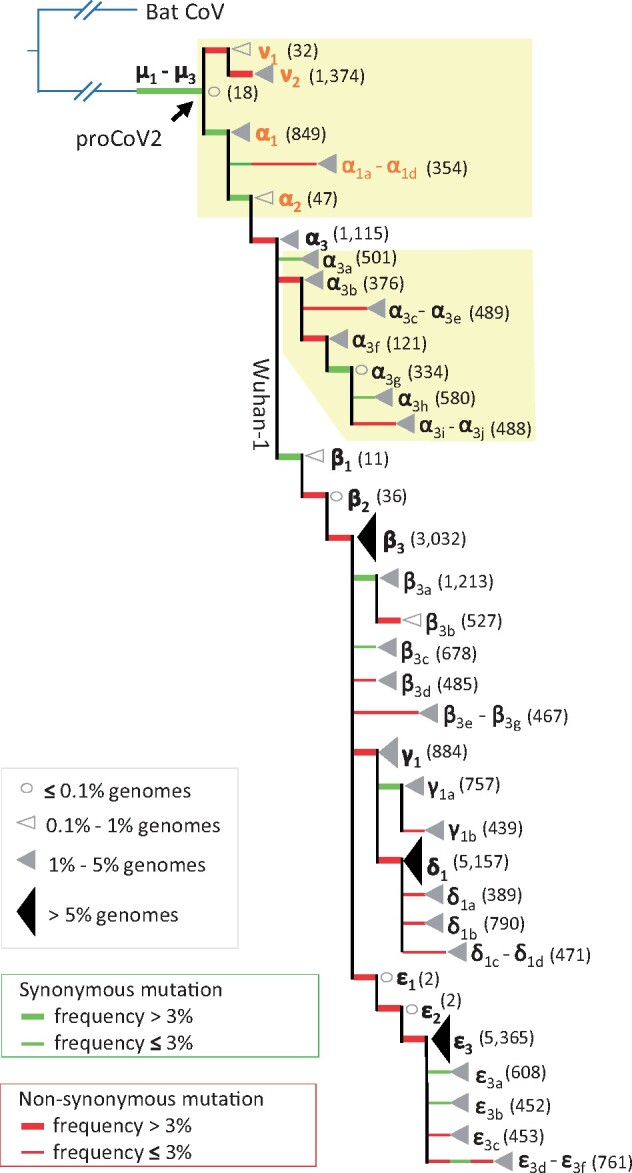
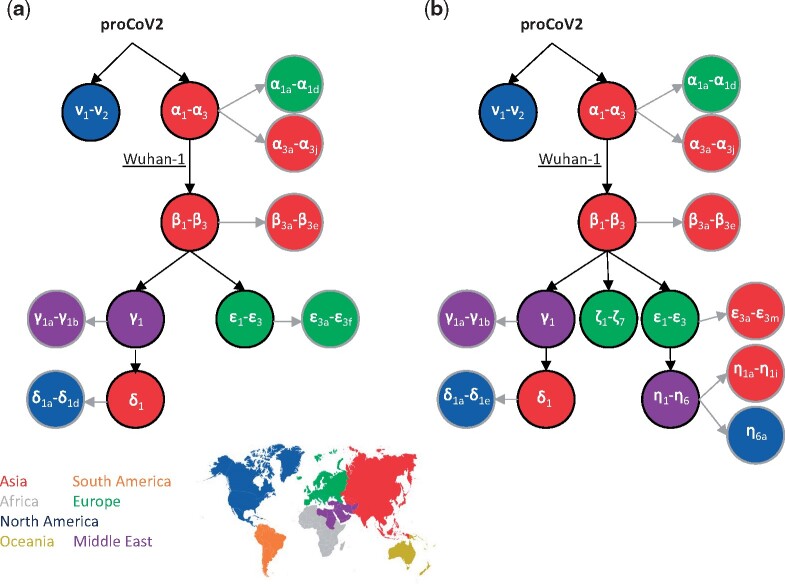
Update of
-
An evolutionary portrait of the progenitor SARS-CoV-2 and its dominant offshoots in COVID-19 pandemic.bioRxiv [Preprint]. 2021 Jan 19:2020.09.24.311845. doi: 10.1101/2020.09.24.311845. bioRxiv. 2021. Update in: Mol Biol Evol. 2021 Jul 29;38(8):3046-3059. doi: 10.1093/molbev/msab118. PMID: 32995781 Free PMC article. Updated. Preprint.
References
-
- Boni MF, Lemey P, Jiang X, Lam TTY, Perry BW, Castoe TA, Rambaut A, Robertson DL.. 2020. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 5(11):1408–1417. - PubMed
-
- Casals F, Bertranpetit J.. 2012. Human genetic variation, shared and private. Science 337(6090):39–40. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
