

Rampant Genome-Wide Admixture across the Heliconius Radiation
- PMID: 33944917
- PMCID: PMC8283734
- DOI: 10.1093/gbe/evab099
Rampant Genome-Wide Admixture across the Heliconius Radiation
Abstract
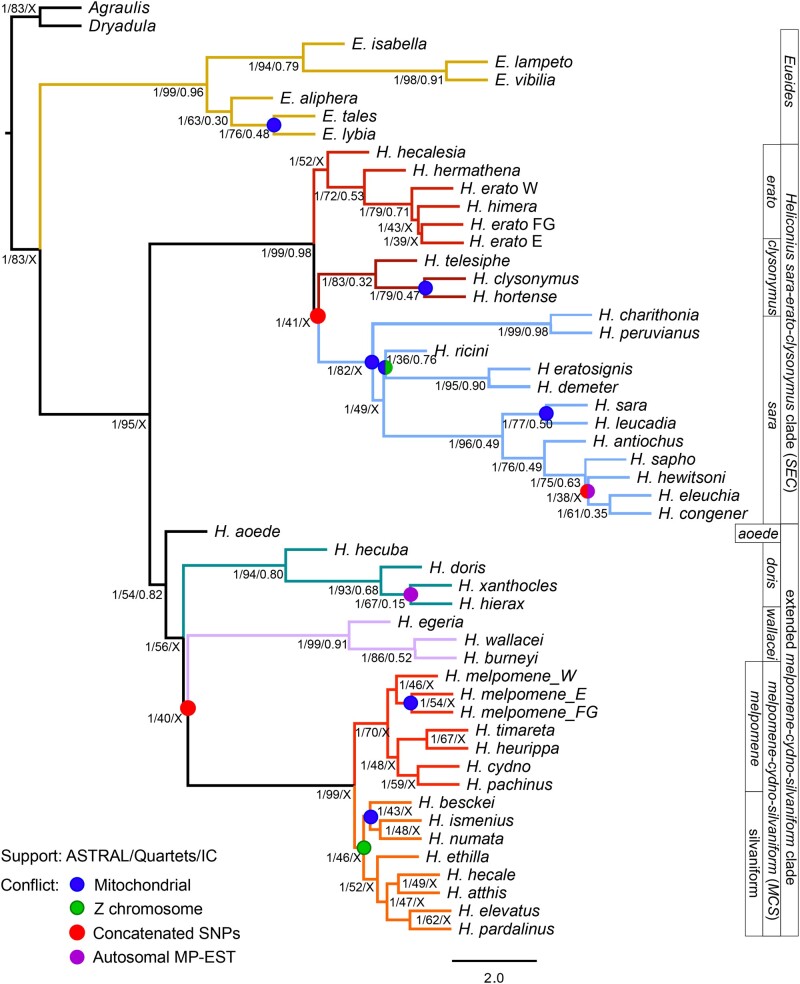
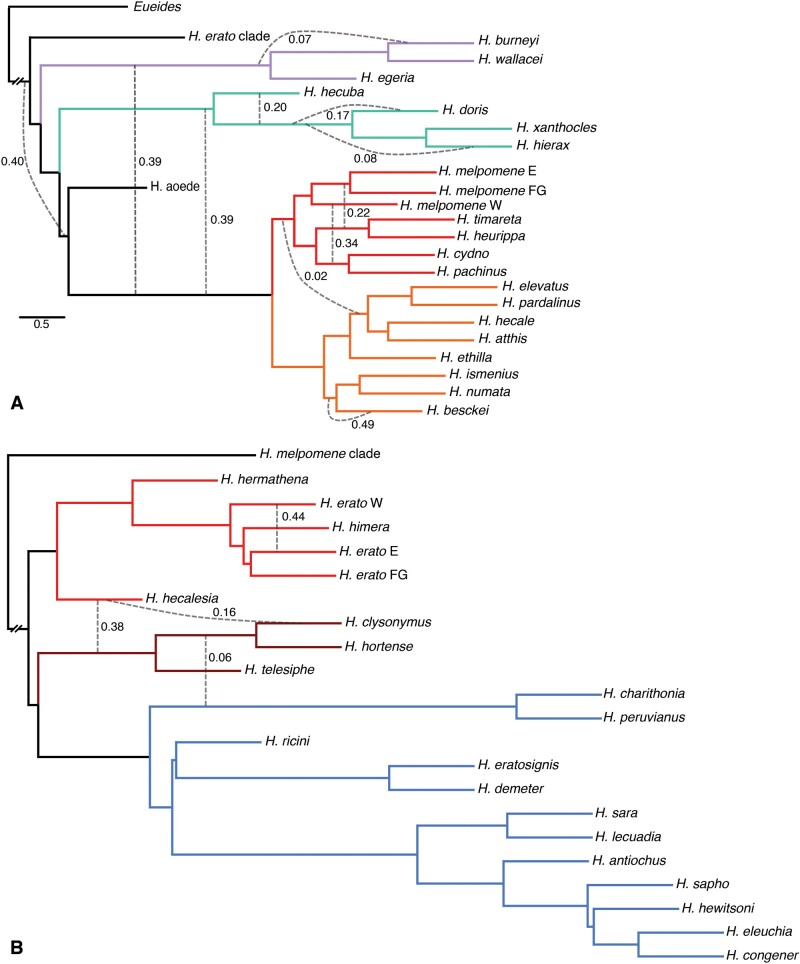
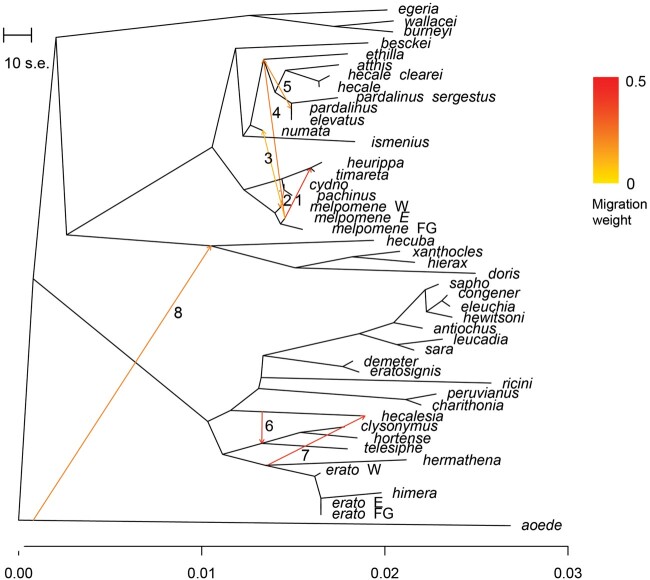
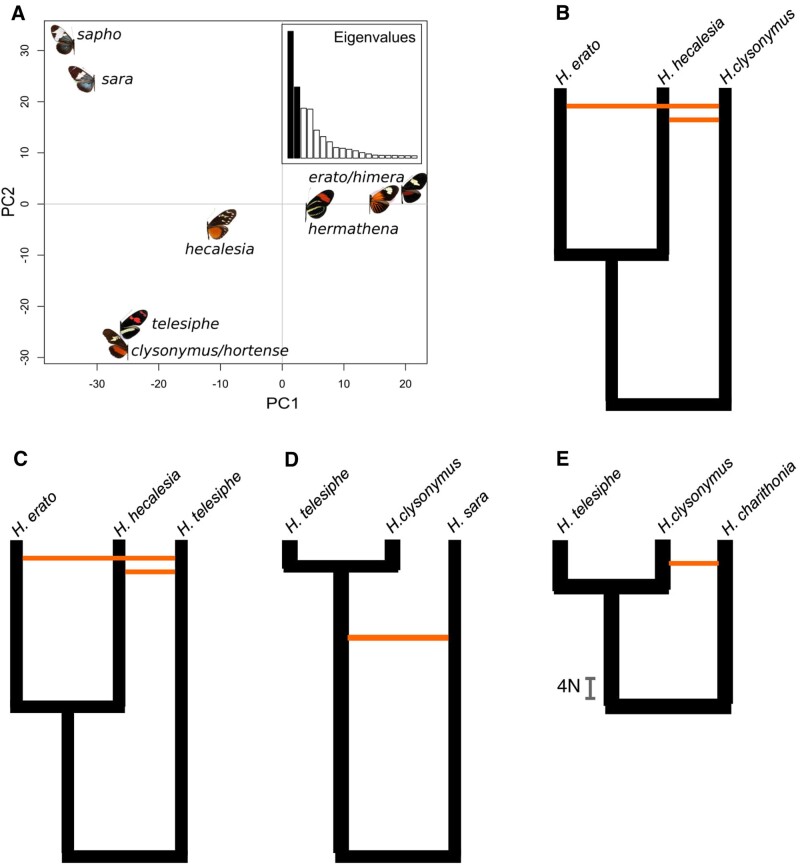
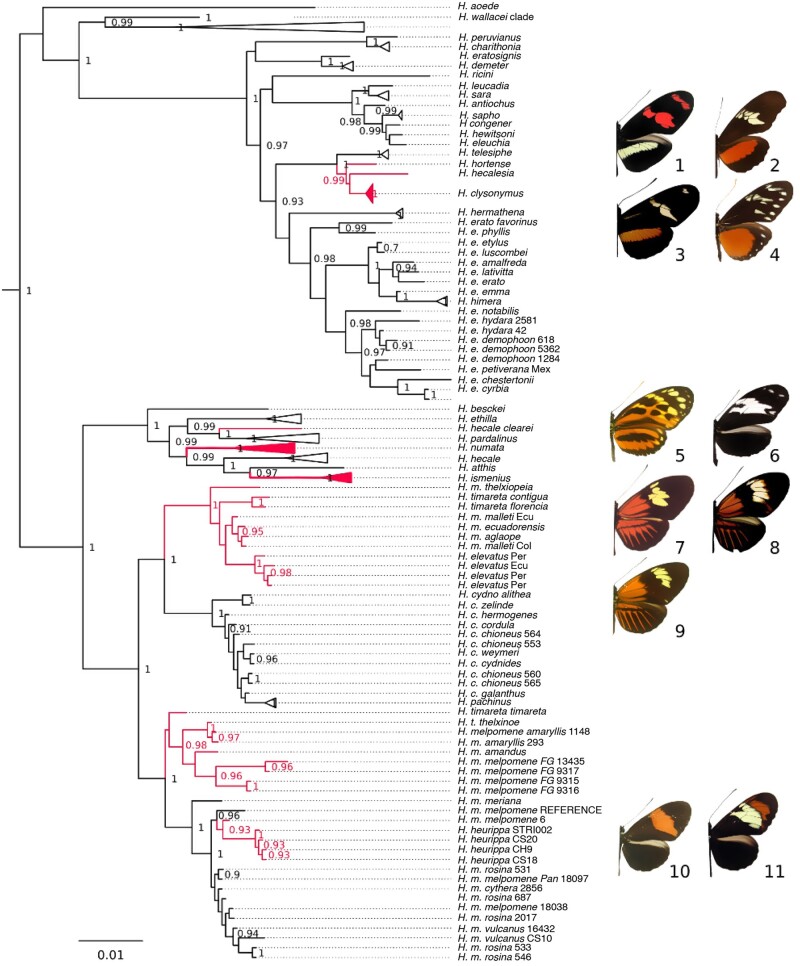
How frequent is gene flow between species? The pattern of evolution is typically portrayed as a phylogenetic tree, yet gene flow between good species may be an important mechanism in diversification, spreading adaptive traits and leading to a complex pattern of phylogenetic incongruence. This process has thus far been studied mainly among a few closely related species, or in geographically restricted areas such as islands, but not on the scale of a continental radiation. Using a genomic representation of 40 out of 47 species in the genus, we demonstrate that admixture has played a role throughout the evolution of the charismatic Neotropical butterflies Heliconius. Modeling of phylogenetic networks based on the exome uncovers up to 13 instances of interspecific gene flow. Admixture is detected among the relatives of Heliconius erato, as well as between the ancient lineages leading to modern clades. Interspecific gene flow played a role throughout the evolution of the genus, although the process has been most frequent in the clade of Heliconius melpomene and relatives. We identify Heliconius hecalesia and relatives as putative hybrids, including new evidence for introgression at the loci controlling the mimetic wing patterns. Models accounting for interspecific gene flow yield a more complete picture of the radiation as a network, which will improve our ability to study trait evolution in a realistic comparative framework.
Keywords: adaptive introgression; admixture; phylogenomics; radiation.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Andrews S. 2014. FastQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [accessed 2014 July 1]
-
- Anisimova M, Gascuel O. 2006. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 55(4):539–552. - PubMed
-
- Bastide P, Solís-Lemus C, Kriebel R, William Sparks K, Ané C. 2018. Phylogenetic comparative methods on phylogenetic networks with reticulations. Syst Biol. 67(5):800–820. - PubMed
-
- Beltran M, et al. 2007. Do pollen feeding, pupal-mating and larval gregariousness have a single origin in Heliconius butterflies? Inferences from multilocus DNA sequence data. Proc R Soc B. 92:221–239.
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
