

Autism-associated SHANK3 missense point mutations impact conformational fluctuations and protein turnover at synapses
- PMID: 33945465
- PMCID: PMC8169116
- DOI: 10.7554/eLife.66165
Autism-associated SHANK3 missense point mutations impact conformational fluctuations and protein turnover at synapses
Abstract
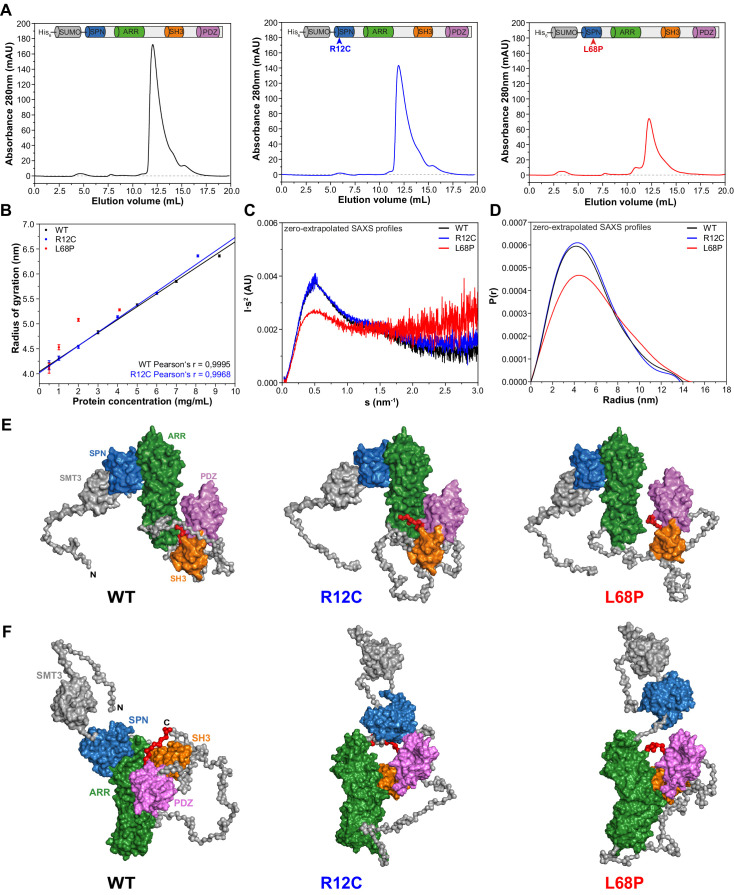
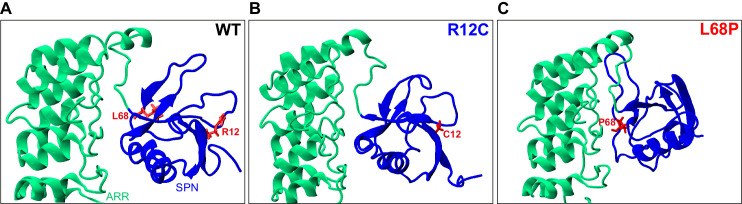
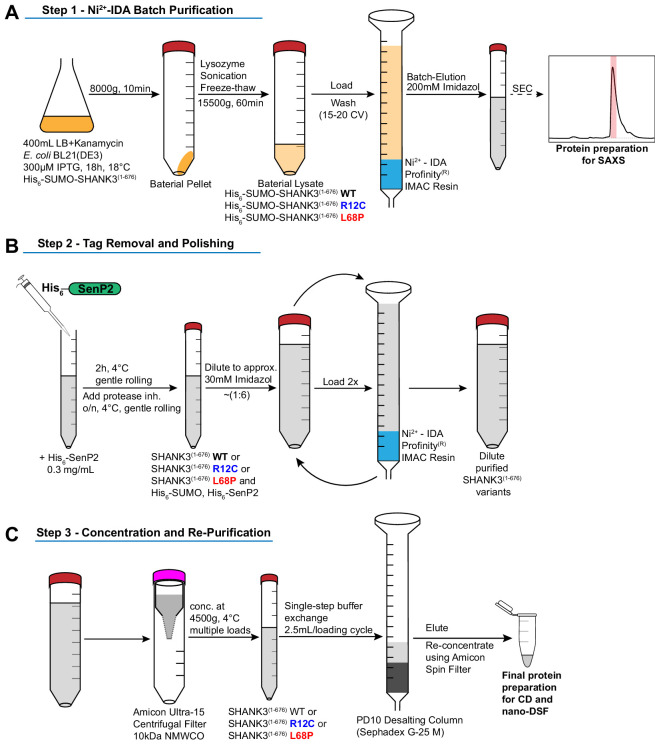
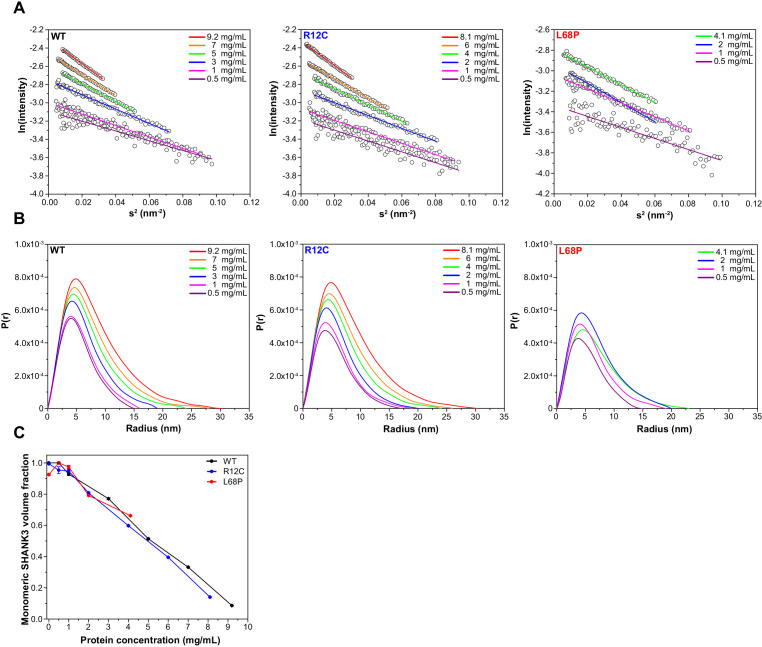
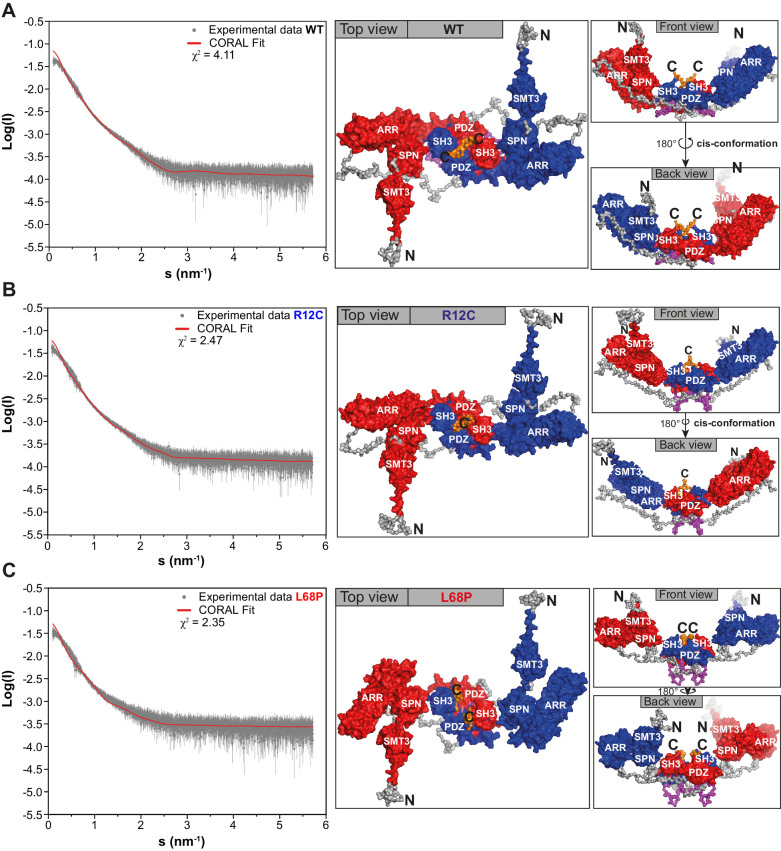
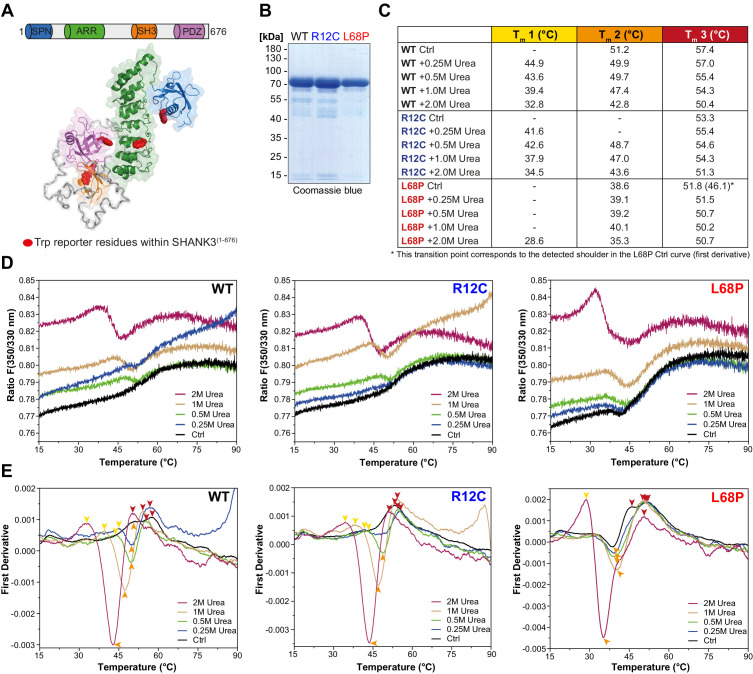
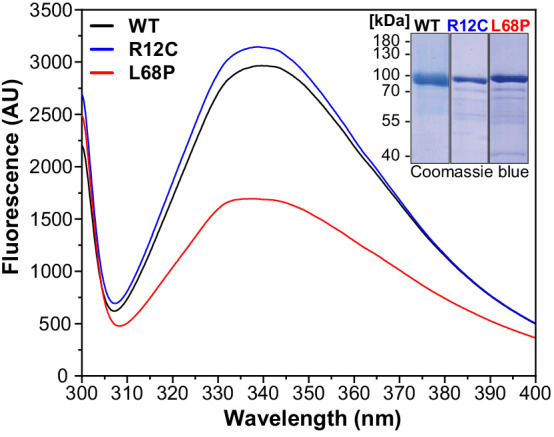
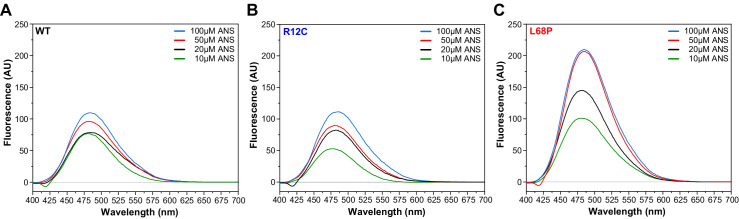
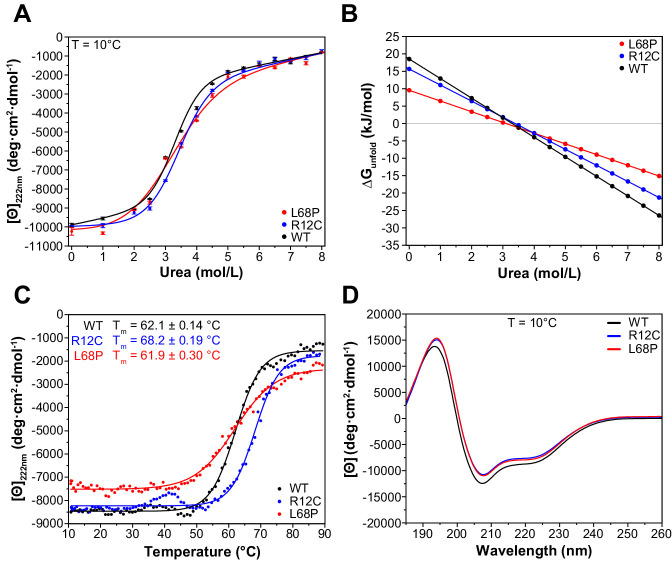
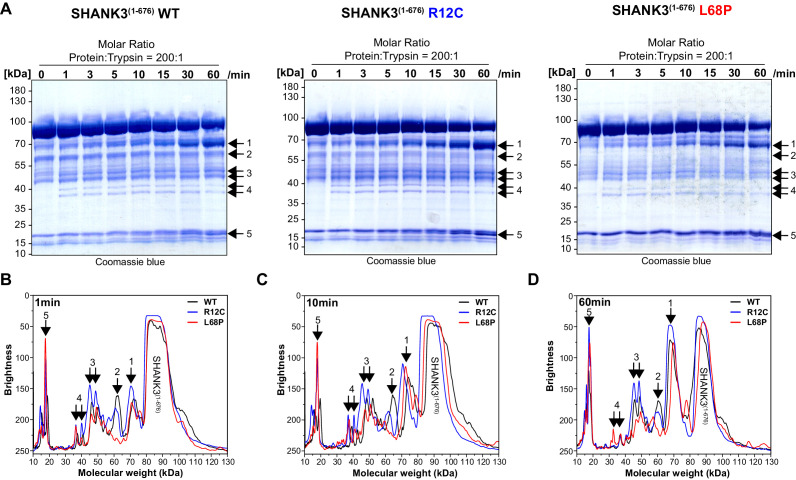
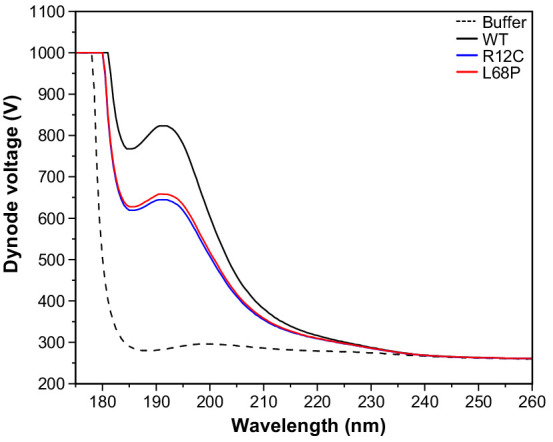
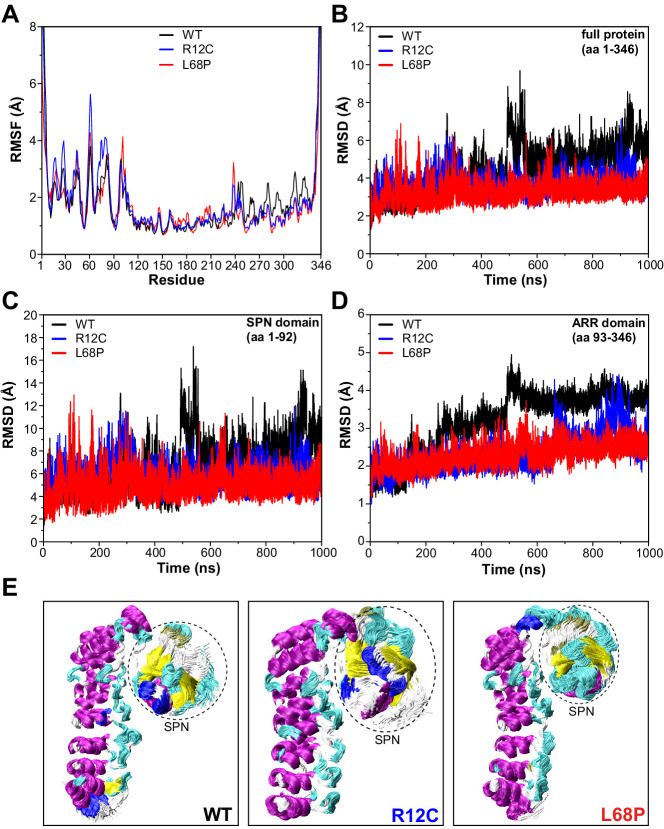
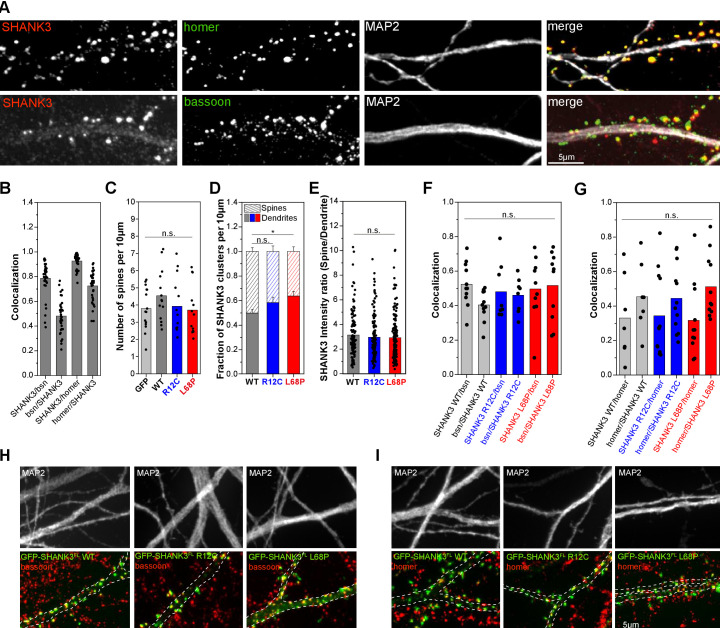
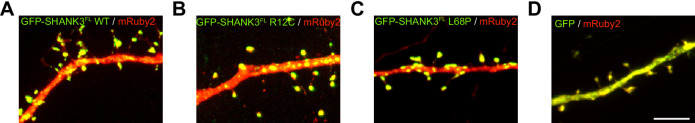
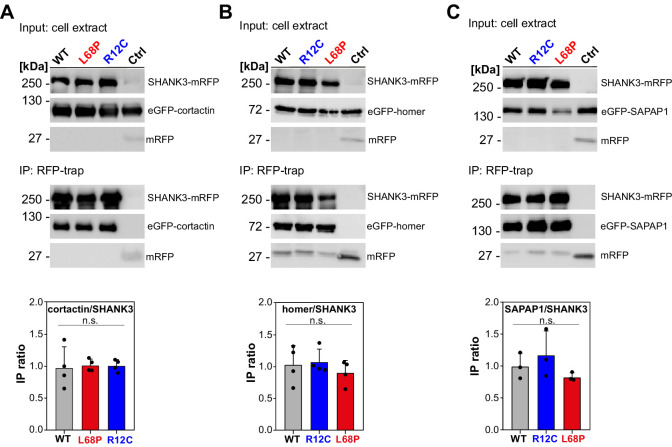
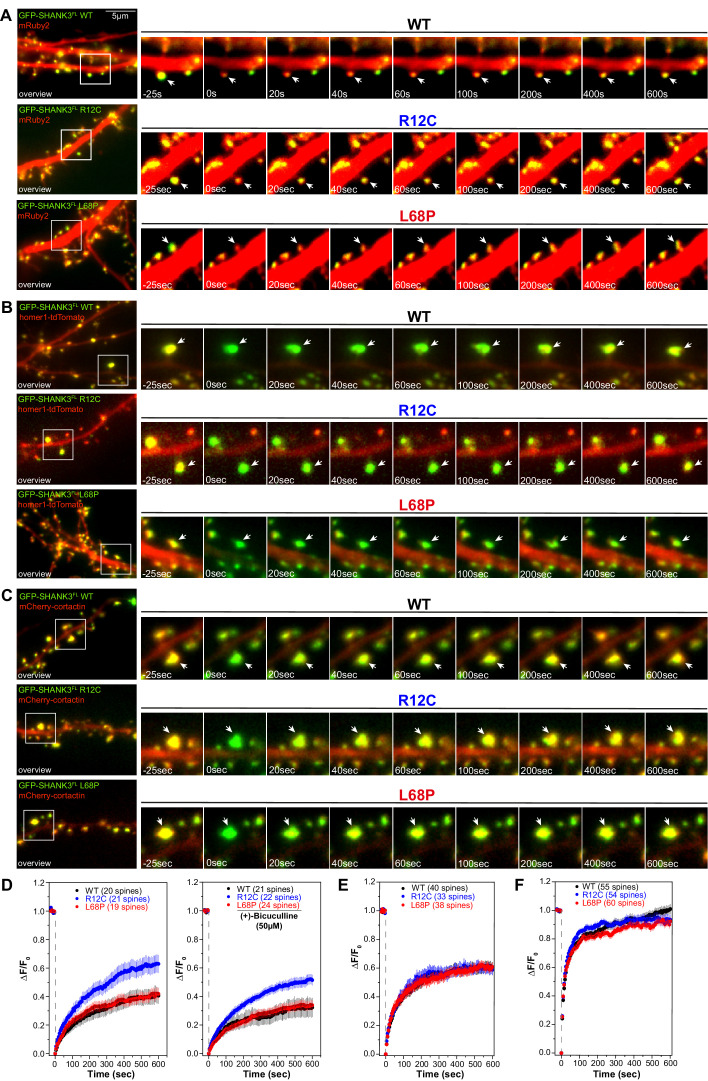
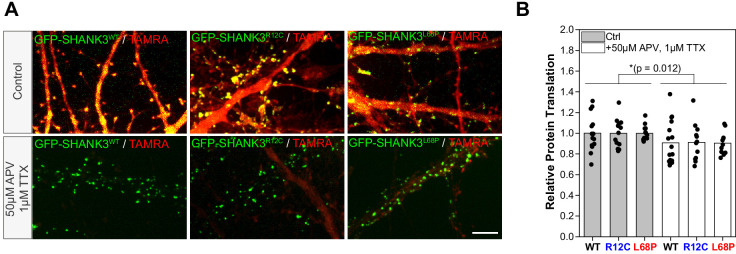
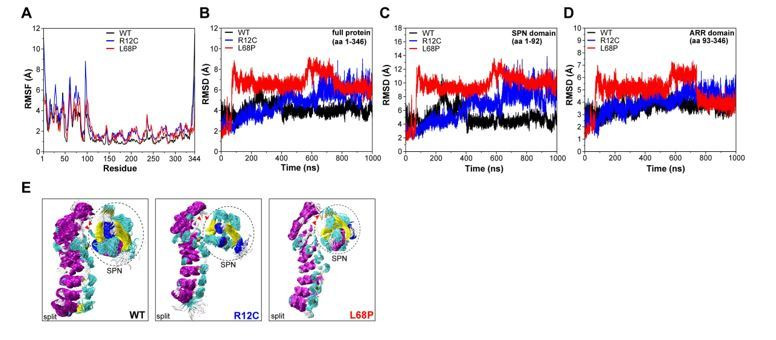
Members of the SH3- and ankyrin repeat (SHANK) protein family are considered as master scaffolds of the postsynaptic density of glutamatergic synapses. Several missense mutations within the canonical SHANK3 isoform have been proposed as causative for the development of autism spectrum disorders (ASDs). However, there is a surprising paucity of data linking missense mutation-induced changes in protein structure and dynamics to the occurrence of ASD-related synaptic phenotypes. In this proof-of-principle study, we focus on two ASD-associated point mutations, both located within the same domain of SHANK3 and demonstrate that both mutant proteins indeed show distinct changes in secondary and tertiary structure as well as higher conformational fluctuations. Local and distal structural disturbances result in altered synaptic targeting and changes of protein turnover at synaptic sites in rat primary hippocampal neurons.
Keywords: SHANK3; autism; conformational dynamics; neuroscience; postsynaptic density; protein folding; rat; synaptic protein turnover.
© 2021, Bucher et al.
Conflict of interest statement
MB, SN, YH, DM, FH, HK, DS, EK, AK, MK, MM No competing interests declared
Figures


References
-
- Alexander CG, Wanner R, Johnson CM, Breitsprecher D, Winter G, Duhr S, Baaske P, Ferguson N. Novel microscale approaches for easy, rapid determination of protein stability in academic and commercial settings. Biochimica Et Biophysica Acta (BBA) - Proteins and Proteomics. 2014;1844:2241–2250. doi: 10.1016/j.bbapap.2014.09.016. - DOI - PMC - PubMed
-
- Blanchet CE, Spilotros A, Schwemmer F, Graewert MA, Kikhney A, Jeffries CM, Franke D, Mark D, Zengerle R, Cipriani F, Fiedler S, Roessle M, Svergun DI. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY) Journal of Applied Crystallography. 2015;48:431–443. doi: 10.1107/S160057671500254X. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
