

Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation
- PMID: 33946927
- PMCID: PMC8146072
- DOI: 10.3390/cells10051056
Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation
Erratum in
-
Correction: Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056.Cells. 2022 Nov 14;11(22):3593. doi: 10.3390/cells11223593. Cells. 2022. PMID: 36429135 Free PMC article.
Abstract
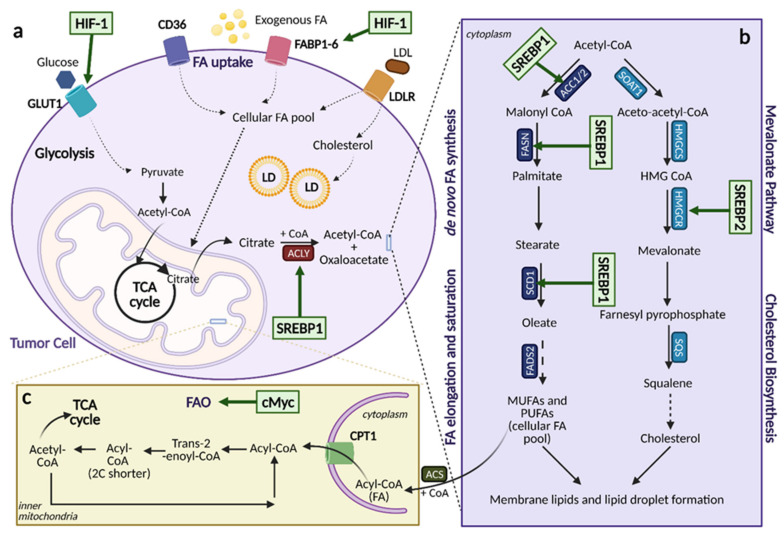
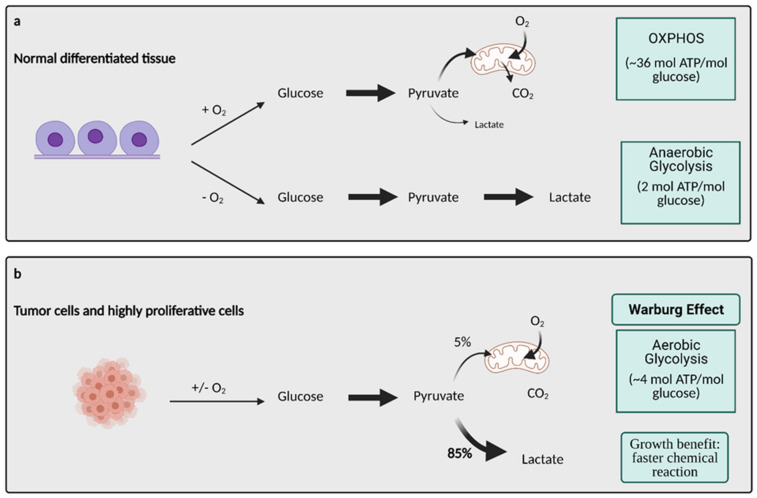
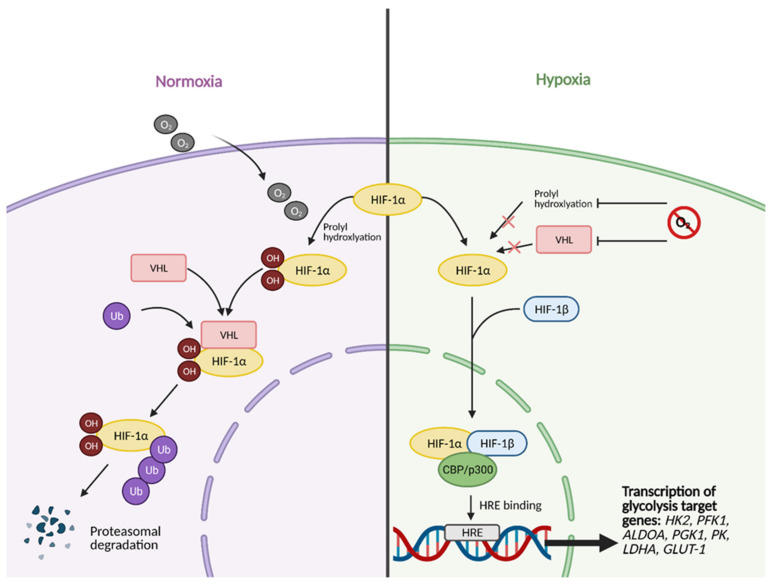
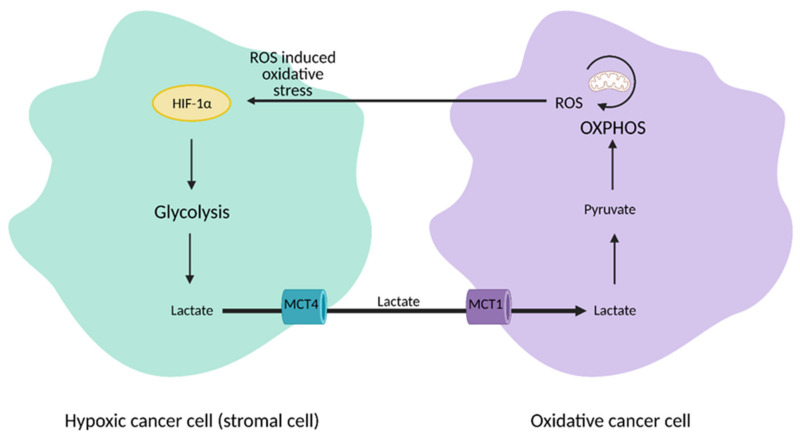
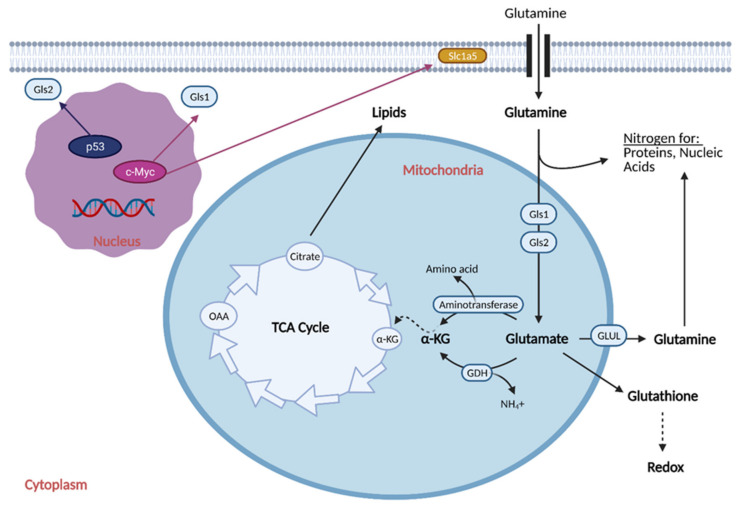
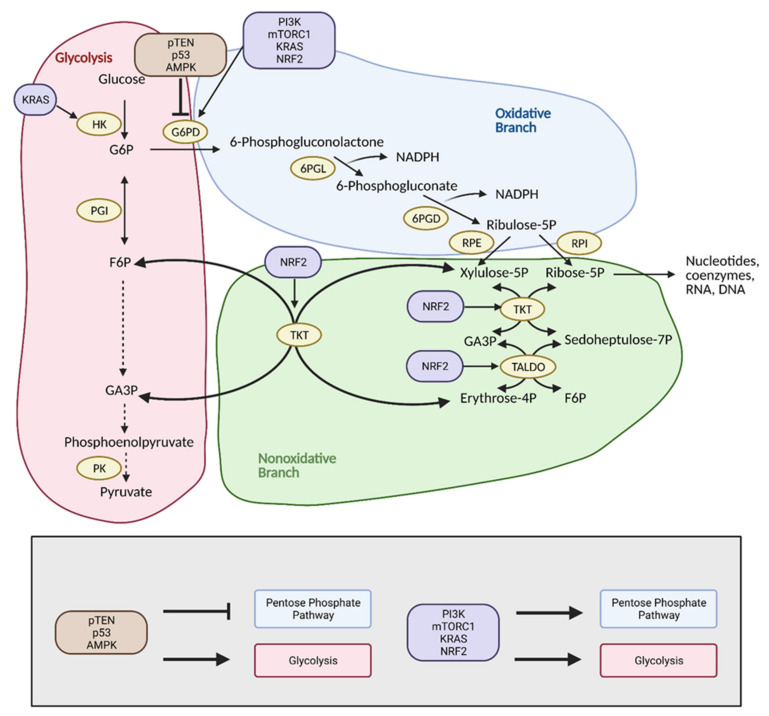
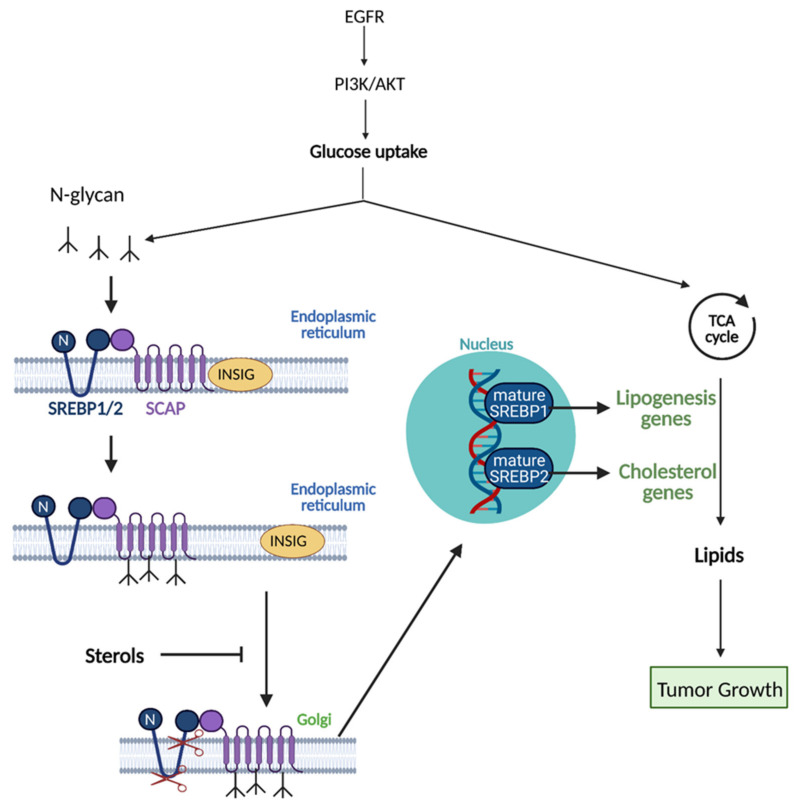
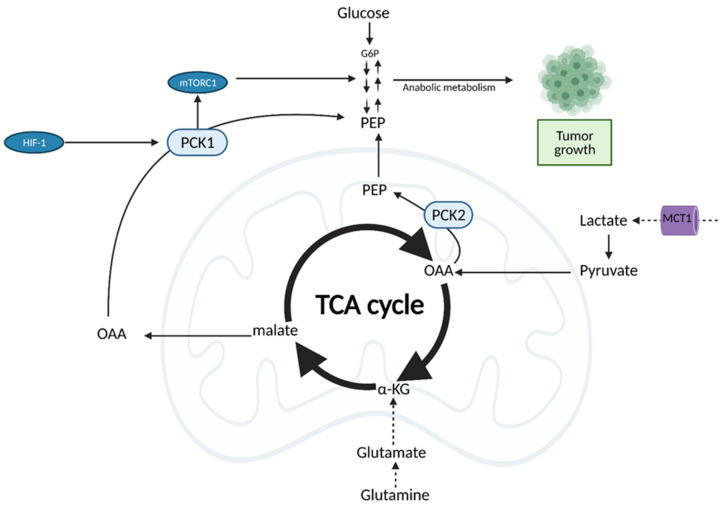
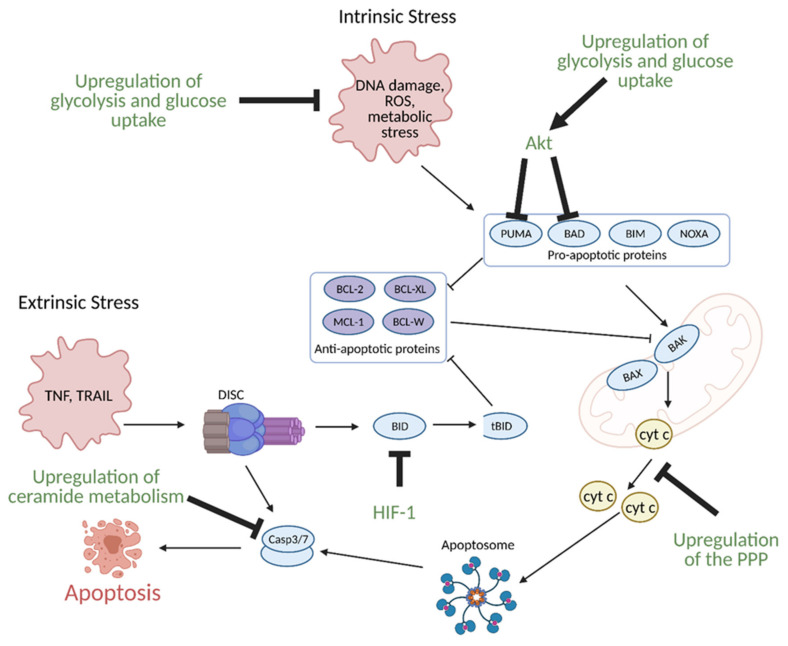
Cancer cells alter metabolic processes to sustain their characteristic uncontrolled growth and proliferation. These metabolic alterations include (1) a shift from oxidative phosphorylation to aerobic glycolysis to support the increased need for ATP, (2) increased glutaminolysis for NADPH regeneration, (3) altered flux through the pentose phosphate pathway and the tricarboxylic acid cycle for macromolecule generation, (4) increased lipid uptake, lipogenesis, and cholesterol synthesis, (5) upregulation of one-carbon metabolism for the production of ATP, NADH/NADPH, nucleotides, and glutathione, (6) altered amino acid metabolism, (7) metabolism-based regulation of apoptosis, and (8) the utilization of alternative substrates, such as lactate and acetate. Altered metabolic flux in cancer is controlled by tumor-host cell interactions, key oncogenes, tumor suppressors, and other regulatory molecules, including non-coding RNAs. Changes to metabolic pathways in cancer are dynamic, exhibit plasticity, and are often dependent on the type of tumor and the tumor microenvironment, leading in a shift of thought from the Warburg Effect and the "reverse Warburg Effect" to metabolic plasticity. Understanding the complex nature of altered flux through these multiple pathways in cancer cells can support the development of new therapies.
Keywords: Warburg Effect; aerobic glycolysis; cancer; one-carbon metabolism; oxidative phosphorylation; pentose phosphate pathway.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Warburg O. The metabolism of carcinoma cells. J. Cancer Res. 1925;9:148–163. doi: 10.1158/jcr.1925.148. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
