

Hemistepsin a Induces Apoptosis of Hepatocellular Carcinoma Cells by Downregulating STAT3
- PMID: 33947048
- PMCID: PMC8125382
- DOI: 10.3390/ijms22094743
Hemistepsin a Induces Apoptosis of Hepatocellular Carcinoma Cells by Downregulating STAT3
Abstract
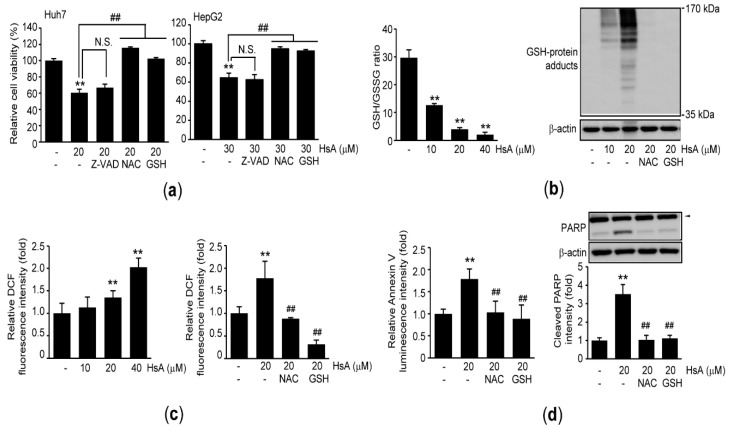
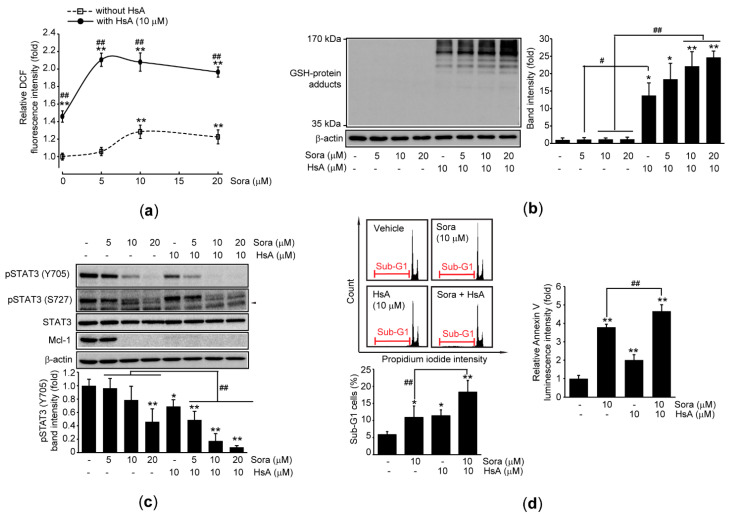
Hemistepta lyrata (Bunge) Bunge is a biennial medicinal plant possessing beneficial effects including anti-inflammation, and hemistepsin A (HsA) isolated from H. lyrata has been known as a hepatoprotective sesquiterpene lactone. In this report, we explored the cytotoxic effects of H. lyrata on hepatocellular carcinoma (HCC) cells and investigated the associated bioactive compounds and their relevant mechanisms. From the viability results of HCC cells treated with various H. lyrata extracts, HsA was identified as the major compound contributing to the H. lyrata-mediated cytotoxicity. HsA increased expression of cleaved PARP and cells with Sub-G1 phase, Annexin V binding, and TUNEL staining, which imply HsA induces apoptosis. In addition, HsA provoked oxidative stress by decreasing the reduced glutathione/oxidized glutathione ratio and accumulating reactive oxygen species and glutathione-protein adducts. Moreover, HsA inhibited the transactivation of signal transducer and activator of transcription 3 (STAT3) by its dephosphorylation at Y705 and glutathione conjugation. Stable expression of a constitutive active mutant of STAT3 prevented the reduction of cell viability by HsA. Finally, HsA enhanced the sensitivity of sorafenib-mediated cytotoxicity by exaggerating oxidative stress and Y705 dephosphorylation of STAT3. Therefore, HsA will be a promising candidate to induce apoptosis of HCC cells via downregulating STAT3 and sensitizing conventional chemotherapeutic agents.
Keywords: Hemistepta lyrata (Bunge) Bunge; apoptosis; hemistepsin A (HsA); hepatocellular carcinoma (HCC) cells; oxidative stress; signal transducer and activator of transcription 3 (STAT3); sorafenib.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- National Cancer Information Center. [(accessed on 30 March 2021)]; Available online: https://www.cancer.go.kr.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
