

Mitochondrial abnormalities: a hub in metabolic syndrome-related cardiac dysfunction caused by oxidative stress
- PMID: 33950478
- PMCID: PMC9197868
- DOI: 10.1007/s10741-021-10109-6
Mitochondrial abnormalities: a hub in metabolic syndrome-related cardiac dysfunction caused by oxidative stress
Abstract
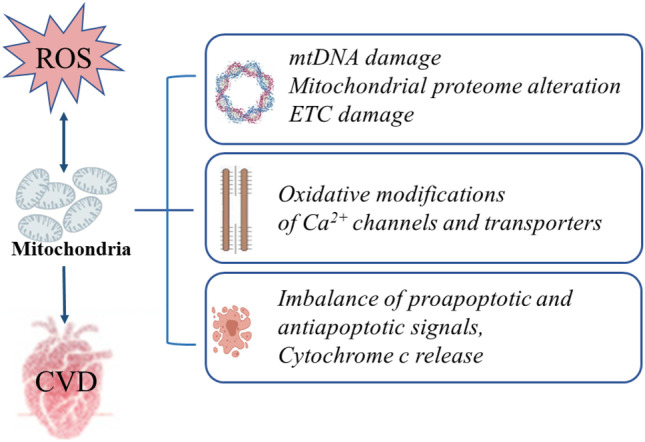
Metabolic syndrome (MetS) refers to a group of cardiovascular risk elements comprising insulin resistance, obesity, dyslipidemia, increased glucose intolerance, and increased blood pressure. Individually, all the MetS components can lead to cardiac dysfunction, while their combination generates additional risks of morbidity and mortality. Growing evidence suggests that oxidative stress, a dominant event in cellular damage and impairment, plays an indispensable role in cardiac dysfunction in MetS. Oxidative stress can not only disrupt mitochondrial activity through inducing oxidative damage to mitochondrial DNA, RNA, lipids, and proteins but can also impair cardiomyocyte contractile function via mitochondria-related oxidative modifications of proteins central to excitation-contraction coupling. Furthermore, excessive reactive oxygen species (ROS) generation can lead to the activation of several mitochondria apoptotic signaling pathways, release of cytochrome c, and eventual induction of myocardial apoptosis. This review will focus on such processes of mitochondrial abnormalities in oxidative stress induced cardiac dysfunction in MetS.
Keywords: Cardiac dysfunction; Metabolic syndrome; Mitochondria; Oxidative stress; Reactive oxygen species.
© 2021. The Author(s).
Conflict of interest statement
All authors declare no competing interests.
Figures
References
-
- Wei YH, Lu CY, Wei CY, et al. Oxidative stress in human aging and mitochondrial disease-consequences of defective mitochondrial respiration and impaired antioxidant enzyme system. Chin J Physiol. 2001;44:1–11. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
