

A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast
- PMID: 33950524
- PMCID: PMC8167363
- DOI: 10.15252/embj.2020107333
A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast
Abstract
To examine global changes in breast heterogeneity across different states, we determined the single-cell transcriptomes of > 340,000 cells encompassing normal breast, preneoplastic BRCA1+/- tissue, the major breast cancer subtypes, and pairs of tumors and involved lymph nodes. Elucidation of the normal breast microenvironment revealed striking changes in the stroma of post-menopausal women. Single-cell profiling of 34 treatment-naive primary tumors, including estrogen receptor (ER)+ , HER2+ , and triple-negative breast cancers, revealed comparable diversity among cancer cells and a discrete subset of cycling cells. The transcriptomes of preneoplastic BRCA1+/- tissue versus tumors highlighted global changes in the immune microenvironment. Within the tumor immune landscape, proliferative CD8+ T cells characterized triple-negative and HER2+ cancers but not ER+ tumors, while all subtypes comprised cycling tumor-associated macrophages, thus invoking potentially different immunotherapy targets. Copy number analysis of paired ER+ tumors and lymph nodes indicated seeding by genetically distinct clones or mass migration of primary tumor cells into axillary lymph nodes. This large-scale integration of patient samples provides a high-resolution map of cell diversity in normal and cancerous human breast.
Keywords: BRCA1 carriers; LN metastasis; breast cancer; microenvironment; single-cell RNA-seq.
© 2021 The Authors. Published under the terms of the CC BY NC ND 4.0 license.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

Schematic diagram showing workflow for scRNA‐seq of human specimens: normal and preneoplastic breast tissue, breast tumors (TNBC, ER+, HER2+, male breast tumors), and matching pairs of tumor and lymph node (LN) samples.
Flow cytometry based on CD49f and EpCAM staining separates lineage‐negative breast tissue cells into stromal (CD49f–EpCAM–) and epithelial cells, which includes basal (CD49f+EpCAMlo/–), luminal progenitor (LP) (CD49f+EpCAM+), and mature luminal (ML) (CD49f–EpCAM+) cells.
t‐SNE plot of the integrated scRNA‐seq profiles of epithelial cells from 11 reduction mammoplasties. Cell colors correspond to tissue specimens.
Same t‐SNE plot as (C) but separated by hormonal status of the donor (8 premenopausal and 3 post‐menopausal).
Same t‐SNE plot as (C) but colored by cell clusters (with Seurat cluster resolution set to 0.015).
Ternary plot positioning each cell according to the proportion of basal, LP, or ML signature genes expressed by that cell. The three vertices of the plot correspond to cells expressing basal genes only, LP genes only, or ML genes only. Cells expressing equal numbers of basal, LP, and ML genes are in the center of the plot. The plot shows the same cells and cell colors as for (E), thus identifying green as basal, blue as LP, and orange as ML populations, respectively.
Diffusion map of the epithelial cells.
t‐SNE plot as in (E) colored by the expression level of a selection of basal, LP, and ML marker genes. Red=high expression, gray=not‐detected.
The basal cell cluster shows a “tail” of atypical cells (highlighted in pink). These tail cells are in the center of the diffusion plot, downstream of the other basal cells in pseudo‐time and intermediate between the basal and two luminal lineages.

Scatterplot showing the number of cells and the number of genes expressed after quality checking and filtering for each of the 79 samples and 10x runs listed in Table EV4.
Flow cytometric analysis of pre‐ and post‐menopausal tissue using CD49f and EpCAM to fractionate lineage‐negative breast cells into epithelial and stromal cells. Cell clusters corresponding to stromal, basal, LP, and ML cell populations were visible for each tissue specimen, but the percentage of cells in each population varied between samples (pre‐ vs post‐menopausal).
t‐SNE plot of the integrated scRNA‐seq profiles of epithelial cells from 11 reduction mammoplasties colored by tissue specimen (left) or by cell cluster (right, with Seurat cluster resolution set to 0.015). Cluster 4 (red) corresponds to stromal cells.
Box plots of lineage‐specific expression signatures by epithelial cell cluster. Each epithelial cell was interrogated with the expression signatures for the human basal, LP, ML, and stromal cell types. Each panel shows the same cells and cell colors as in Fig 1E. Labels 1–4 correspond to clusters in Fig 1E. The four panels show basal, LP, ML, and stromal expression signatures, respectively. Vertical axis shows average expression of cell population signature genes as log2 counts per million. Box plots show quartiles, minimum, and maximum.
Heat map of pseudo‐bulk samples showing marker genes for each epithelial cluster. The top 20 marker genes were identified for each cluster by differential expression analysis of the pseudo‐bulk read counts. Color bars at the top of the plot indicate the cluster and patient sample.
Same t‐SNE plot as Fig 2A and B showing combined profiles of total tissue cells from reduction mammoplasties but colored by epithelial lineage expression scores. Expression levels confirm the green, blue, and red clusters as basal, LP, and ML, respectively.
As for (F) but colored by expression of EMT marker genes (ZEB1/2, SNAI1).
Heat map of non‐epithelial pseudo‐bulk samples showing expression of immune and stromal lineage genes from Novershtern et al (2011) and Jeffrey et al (2006). Sample colors correspond to cell clusters in Fig 2D.

t‐SNE map of combined scRNA‐seq transcriptomes of total tissue cells from 13 reduction mammoplasties. Cell colors correspond to tissue specimens.
Same t‐SNE map as (A) but colored by cell cluster (with cluster resolution 0.05).
Ternary plot positioning each cell according to the proportion of basal, LP, or ML signature genes expressed by that cell. The plot shows the same cells and cell colors as for (B).
Reclustered EPCAM‐negative non‐epithelial cells revealed seven clusters (resolution 0.05) representing immune and stromal cell lineages.
Multidimensional scaling plot showing expression profile distances between the pseudo‐bulk samples. Each dot corresponds to aggregated expression for a cell cluster for one patient. Cluster colors are overlaid from (D, right panel). Distances on the plot correspond to leading log2‐fold change, defined as the average log2‐fold change for the 500 most differential genes between each pair of profiles.
Heat map of pseudo‐bulk samples showing marker genes for each non‐epithelial cluster. The top 20 marker genes were identified for each cluster by differential expression analysis of the pseudo‐bulk read counts. Color bars at the top of the plot indicate the cluster and menopausal status (blue/yellow for pre/post‐menopause) of each sample.
Same t‐SNE map of non‐epithelial cells as in (D) but colored by menopausal status. Barplot shows relative cluster sizes (percentage of total cells) for each status condition. Clusters 1 and 2 have significantly different sizes in post‐menopause samples after allowing for inter‐patient variability (P = 0.040 and P = 0.032 by quasi‐binomial F‐test). Sizes are not significantly different for other clusters (P > 0.15).
Relative cluster sizes as for (G) but by individual patients. Cluster colors correspond to (D) by cluster. A quasi‐multinomial F‐test was used to test for differences in cluster frequencies between pre‐ and post‐menopausal samples (P = 0.007).
Bar plots showing percentage of cells expressing selected immune and endothelial cell markers for pre‐ and post‐menopausal samples and the cell clusters identified in Fig 2D.

Microenvironment t‐SNE map as in Fig 2D and G but separated by menopausal status and colored by expression (red=high expression, gray=undetectable) of selected fibroblast markers (upper panel). The dotted lines indicate the pericyte subsets. Bottom panel of bar plots shows percentage of cells expressing the markers for the cell clusters identified in Fig 2D.
Co‐immunofluorescence staining of pre‐ versus post‐menopausal tissue for E‐cadherin (cyan), PDGFRβ (yellow), and F‐actin (pink). DAPI is shown in gray. The arrowheads in (v) depict fibroblasts in direct contact with the myoepithelial layer. For 2D and 3D confocal imaging: n = 13 premenopausal and n = 10 post‐menopausal specimens. Scale bars: wholemount and optical sections: 100 μm (panels i‐iv); enlargements, 30 μm (panel v).
t‐SNE plot of the integrated scRNA‐seq profiles of fibroblasts from pre‐ and post‐menopausal tissue (reclustered cells from cluster 1 in Fig 2D). Cell colors correspond to tissue specimens.
Same t‐SNE plot as in (C) showing 5 distinct clusters (clusters 3 and 5 were specific to one patient).
KEGG pathways enriched in cluster 1 versus 2 from (D) above (Fisher’s exact test).
Heat map of same cells as in (D) showing expression of the top 20 marker genes in each cluster. Color bars at the top of the plot show cluster membership (colors as in (D)) and pre‐ or post‐menopausal status (blue and yellow, respectively).

t‐SNE map of combined scRNA‐seq profiles of total cells isolated from pathologically normal preneoplastic tissue from BRCA1 mutation carriers (BRCA1; n = 4) and non‐BRCA premenopausal women (n = 8) with no family history of breast cancer. Cell colors represent individual samples.
Same t‐SNE map as in (A) but colored by cluster (cluster resolution 0.12).
Epithelial clusters were identified by EPCAM expression, and the non‐epithelial cells were reclustered to reveal immune and stromal cell populations (cluster resolution 0.08). Lineage identity was determined by hierarchical clustering according to top marker genes (Appendix Fig S1C).
t‐SNE plot showing the combined single‐cell transcriptomes of total tissue cells from BRCA1 preneoplastic tissue and TNBCs (n = 4 for each), colored according to individual patients (B1 = preneoplastic BRCA1+/‐ tissue; TN‐B1= BRCA1‐associated TNBCs).
Same t‐SNE map as (D) but colored by cluster (cluster resolution 0.15).
Expression of epithelial markers indicated on the combined t‐SNE plot as in (D, E) for BRCA1 preneoplastic and BRCA1‐associated tumor cells.
Epithelial clusters were identified by EPCAM expression, and non‐epithelial cells are indicated by the dotted line.
Same t‐SNE map as in (D, E) but colored according to cancerous state: preneoplastic tissue (blue) and BRCA1‐associated TNBCs (yellow).
Box plots of signature expression scores for the 13 cell clusters in (D, E) according to human breast epithelial and stromal gene signatures. Cluster 1 corresponds to tumor cells, while clusters 6, 9, and 10 represent adjacent normal LP, basal, and ML cells, respectively. Box plots show quartiles, minimum, and maximum.
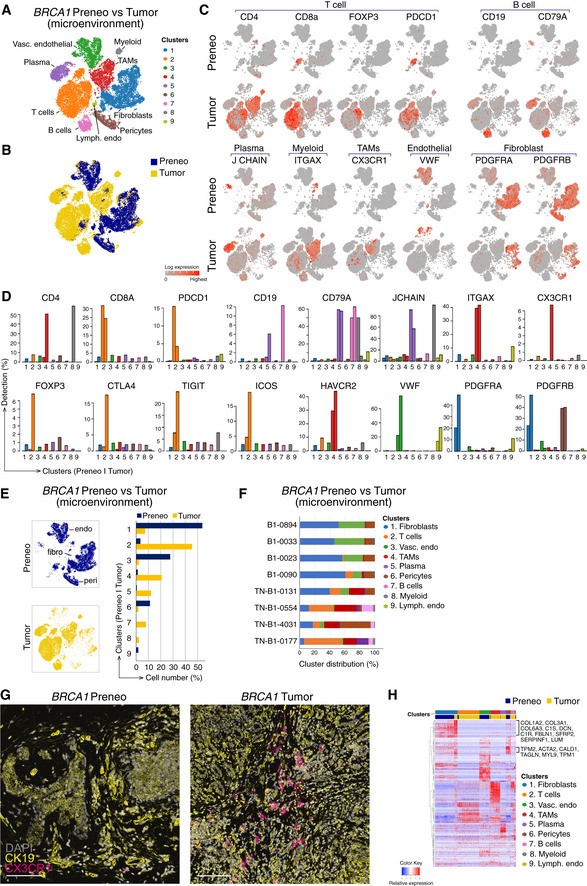
Reclustered EPCAM‐negative cells (excluding clusters 1, 6, 9, and 10 from Fig 4E) revealed immune/stromal cells in the microenvironment, identified using lineage markers.
Combined t‐SNE plot as in (A) of the single‐cell transcriptomes of immune/stromal cells from preneoplastic tissue of BRCA1 mutation carriers (blue; preneo) and BRCA1‐associated TNBCs (yellow; tumor).
t‐SNE plots showing relative expression of cardinal markers of immune and stromal cells in each cell.
Bar plots showing the percentage of cells expressing typical immune cell (including Treg) genes for clusters in (A) by preneoplastic vs tumor.
Left panel, same t‐SNE map as in (A) but separated into cells from preneoplastic (blue) and tumor specimens (yellow). Endothelial cell (endo), fibroblast cell (fibro), and pericytes (peri) clusters are marked. Right panel, proportion of clusters as in (A) according to tissue type.
Relative cluster sizes as in (E) but by individual patient.
Co‐immunofluorescence of tissue stained for the epithelial marker (cytokeratin 19; yellow) and the tumor‐associated macrophage marker CX3CR1 (magenta). DAPI is shown in white (n = 2 preneoplastic samples; n = 2 tumors). Scale bar, 100 μm.
Heat map of top differentially expressed genes in the major immune/stromal cell clusters, identified in (A). BRCA1+/‐ preneoplastic cells, blue; BRCA1‐associated tumor cells, yellow.

- A
Immunostaining of representative ER+, TNBC, and HER2+ primary tumors for expression of ER, PR, HER2, and pan‐cytokeratin. H&E sections are shown in the left panels. Scale bar, 200 μm.
- B–D
Same t‐SNE plots as in Fig 6A–C showing the integrated profiles of total cells from 8 TNBC (4 non‐BRCA1, 4 BRCA1‐mutated), 6 HER2+, and 13 ER+ tumors, respectively. Cells are colored by tissue sample (top panels) or cell cluster (bottom panels). Cluster resolutions: 0.1, 0.07, and 0.1 for TNBC, HER2+, and ER+, respectively.

Box plots showing transcriptional correlations between epithelial subsets (basal, LP, and ML) and cell clusters present in TNBC, HER2+, and ER+ tumors. LP = Luminal progenitor, ML= Mature luminal cells. Cell numbers are given in Fig EV2B–D. Box plots show quartiles, minimum, and maximum.
t‐SNE plots of epithelial cells from TNBC and HER2+ tumors, respectively (EPCAM+ cells in Fig 6B and C). Left panels show reclustering (resolutions 0.1 and 0.05). Right panel shows MKI67 expression.
Same t‐SNE plots as in (B) and Fig 6E, depicting distribution of cells expressing epithelial markers (KRT5 and KRT8), hormone receptors (ESR1, PGR), ERBB2 receptor, EMT signature genes (ZEB1, ZEB2, SNAI1, SNAI2, VIM, and TWIST1) and BCL2.
Enrichment of KEGG pathways in clusters identified from (B) (Fisher’s exact test). For TBNC, cluster 2 vs 1; HER2+, cluster 3 vs the rest.
Box plots of breast cancer subtype signatures from the TCGA by epithelial cell cluster for the combined ER+ tumor dataset. Each panel shows the same cells and cell colors as for the combined dataset in Fig 6E. Vertical axis shows average expression of cell population signature genes as log2 counts per million. Box plots show quartiles, minimum, and maximum.

t‐SNE plots showing T cells (clusters 1 and 5 for TNBC; clusters 2 and 7 for HER2+; cluster 1 for ER+ tumors) identified in Fig 7A–C. Reclustering revealed at least five T‐cell subsets in TNBC and HER2+ tumors, and four T‐cell populations in ER+ tumors (cluster resolutions: 0.2, 0.7, and 0.2, respectively).
Same t‐SNE plots as in (A) but colored by expression of T‐cell markers and of MKI67 to identify proliferating T‐cell clusters.
Heat maps showing pseudo‐bulk expression of top DE genes within the T‐cell subsets identified in the t‐SNE plots shown in (A). TEM = Effector memory T cells; Treg = Regulatory T cells; TRM = Tissue‐resident memory T cells; NK = Natural Killer cells.

Combined t‐SNE transcriptome profiles of two ER+ male tumors; mER‐0068‐T (blue) and mER‐0178T (yellow). Bottom panel shows the corresponding t‐SNE cell clusters, as shown in the top panel, but colored by cell clusters.
t‐SNE plots showing gene expression of epithelial markers and steroid hormone receptors.
Box plots of lineage‐specific expression signatures by epithelial cell cluster. Each epithelial cell was interrogated with the expression signatures for the human basal, LP, ML, and stromal cell types. Each panel shows the same cells and cell colors as in (A). Vertical axis shows average expression of cell population signature genes as log2 counts per million. Box plots show quartiles, minimum, and maximum.
Tumor‐LN pairs: interrogation of cell clusters identified in Fig 9A for expression of canonical immune (CD68, CD19, CD4) and stromal (COL1A2, S100A4, PDGFRA) genes.
Maps of inferred copy number variation (CNV) for the combined tumor‐LN scRNA‐seq expression data for the clusters indicated in Fig 9A. Tumor cells can be distinguished from normal (N) by abundance of CNVs.

- A–C
t‐SNE plots of combined scRNA‐seq profiles of total cells from 8 TNBC tumors, 6 HER2+ tumors, and 13 ER+ tumors, respectively. Integration and cluster sizes for the same cells are shown in Fig EV2, EV3, EV4, EV5. Cells colored by cluster (top left panels), EPCAM expression (top right), cancer subtype marker (bottom left), or MKI67 expression (bottom right). Dotted lines delineate epithelial cells (top panels) and cycling epithelial cells (bottom panels). Normal epithelial subsets (normal) are also demarcated by dotted lines in the upper‐right panels of (B) and (C).
- D
InferCNV plots to map inferred copy number variation (CNV) for the combined tumor scRNA‐seq expression data for the epithelial clusters indicated in panels A‐C. scRNA‐seq data from normal breast epithelial cells (N‐1105‐epi) served as a reference for normalization. Each row represents a gene and each column cells from a cluster in a single tumor. The tumor clusters are color‐coded as in (A‐C). Amplifications (red) and deletions (blue) are inferred from the gene expression. Tumor cells were distinguished from normal (N) cells by abundance of CNV.
- E
t‐SNE plot of EPCAM + epithelial cells from ER+ tumors (C). Top panel shows reclustering (resolution 0.05), bottom panel shows expression of MKI67.
- F
Heat map of cells from clusters in (E) using genes from the PAM50 cancer subtype classifier.
- G
Enrichment of KEGG pathways in EPCAM + clusters 1 to 3 in (E) for ER+ tumors (Fisher’s exact test).
- H
Combined t‐SNE transcriptome profiles of two distinct ER+ tumors isolated from the same breast of a patient: ER‐0029‐7C (blue) and ER‐0029‐9C (yellow). The corresponding t‐SNE cell clusters are shown in the middle panel, and expression of EPCAM is shown in the right‐hand panel.

- A–C
t‐SNE maps of reclustered EPCAM‐negative non‐epithelial cells identified in Fig 6 (A‐C). Cluster resolutions 0.136, 0.1, and 0.1, respectively. The major cell clusters within the microenvironment were identified based on expression of lineage‐specific genes. Heat maps of pseudo‐bulk samples show marker genes for each cluster. The top 30 marker genes were identified for each cluster by differential expression analysis of the pseudo‐bulk read counts. Cluster 9 in HER2+ tumors (B) expressed myeloid and luminal epithelial markers, suggesting phagocytosis of the latter by macrophages. Color bars at the top of the heat map indicate the cluster of each sample; top genes that mark each cluster are indicated.
- D
t‐SNE plots showing the expression of T lymphoid and myeloid markers as shown in (A‐C). Right panels: t‐SNE plots showing the expression of the proliferation marker MKI67 for the same clusters. Dotted lines highlight T and myeloid cells that express MKI67.

- A, B
Image quantification showing number of CD8+Ki67+ cells (A) or CD68+Ki67+ cells (B) per mm2 of tissue from TNBC, HER2+, and ER+ tumors, either within the tumor region (K8/18+) or the stroma (defined as > 5 μm from tumor border). Error bars represent s.e.m. CD8/Ki67: n = 11 for TNBC, n = 12 for ER+, n = 5 for HER2+. CD68/Ki67: n = 6 for TNBC, n = 8 for ER+, n = 5 for HER2+.
- C–F
Representative confocal images of ER+ (ER‐0032) and triple‐negative tumors (TN‐0066) immunolabeled for K8/18 (yellow), CD68 (green) and Ki67 (red) (C, E) or K8/18 (yellow), CD8 (green) and Ki67 (red) (D, F). DAPI is shown in blue. Arrows depict proliferative T cells (CD8+Ki67+) or macrophages (CD68+Ki67+). Enlargements on shown in the right‐hand panels. Scale bars, 200 µm for large tilescans; 50 µm for enlargements and smaller tilescans.

Combined t‐SNE plots of matching tumor and lymph node samples from seven patients (6 female and 1 male) with ER+ disease. Patient 1: ER‐0040‐T and ER‐0040‐LN; Patient 2: ER‐0056‐T and ER‐0056‐LN; Patient 3: ER‐0043‐T and ER‐0043‐LN; Patient 4: ER‐0064‐T and ER‐0064‐LN; Patient 5: ER‐0167‐T and ER‐0167‐LN; Patient 6: ER‐0173‐T and ER‐0173‐LN and Patient 7: mER‐0068‐T and mER‐0068‐LN. The top panels show the combined cells marked according to primary tumor (blue) or involved LN cells (yellow). The middle panels show expression of the epithelial marker EPCAM and proliferation marker MKI67. The bottom panels indicate the major cell clusters and their identity based on expression analyses for lineage‐specific markers.
InferCNV plots were generated from the combined transcriptomes (A) to map copy number variation (CNV) in each chromosome. Tumor cells can be distinguished from normal (N) cells by abundance of CNV.
Immunostaining of tumor and LN from patient ER‐0064 for expression of ER, PR, and pan‐cytokeratin. Insets show sections stained with control isotype antibodies. PR, progesterone receptor. Scale bar, 100 μm.
References
-
- Ali HR, Jackson HW, Zanotelli VRT, Danenberg E, Fischer JR, Bardwell H, Provenzano E, Team CIGC , Rueda OM, Chin SF et al (2020) Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nature Cancer 1: 163–175 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
