

Intergenic RNA mainly derives from nascent transcripts of known genes
- PMID: 33952325
- PMCID: PMC8097831
- DOI: 10.1186/s13059-021-02350-x
Intergenic RNA mainly derives from nascent transcripts of known genes
Abstract
Background: Eukaryotic genomes undergo pervasive transcription, leading to the production of many types of stable and unstable RNAs. Transcription is not restricted to regions with annotated gene features but includes almost any genomic context. Currently, the source and function of most RNAs originating from intergenic regions in the human genome remain unclear.
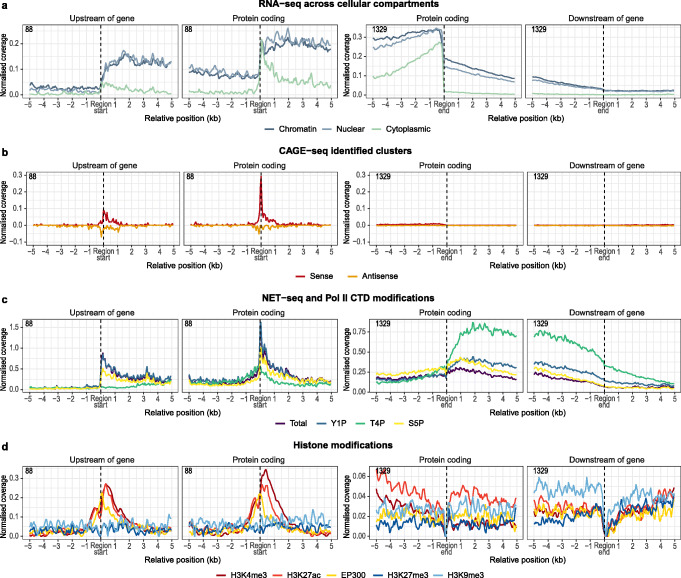
Results: We hypothesize that many intergenic RNAs can be ascribed to the presence of as-yet unannotated genes or the "fuzzy" transcription of known genes that extends beyond the annotated boundaries. To elucidate the contributions of these two sources, we assemble a dataset of more than 2.5 billion publicly available RNA-seq reads across 5 human cell lines and multiple cellular compartments to annotate transcriptional units in the human genome. About 80% of transcripts from unannotated intergenic regions can be attributed to the fuzzy transcription of existing genes; the remaining transcripts originate mainly from putative long non-coding RNA loci that are rarely spliced. We validate the transcriptional activity of these intergenic RNAs using independent measurements, including transcriptional start sites, chromatin signatures, and genomic occupancies of RNA polymerase II in various phosphorylation states. We also analyze the nuclear localization and sensitivities of intergenic transcripts to nucleases to illustrate that they tend to be rapidly degraded either on-chromatin by XRN2 or off-chromatin by the exosome.
Conclusions: We provide a curated atlas of intergenic RNAs that distinguishes between alternative processing of well-annotated genes from independent transcriptional units based on the combined analysis of chromatin signatures, nuclear RNA localization, and degradation pathways.
Keywords: Gene annotation; RNA; RNA-seq; Transcription.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
