

Mutational mechanisms shaping the coding and noncoding genome of germinal center derived B-cell lymphomas
- PMID: 33953289
- PMCID: PMC8257491
- DOI: 10.1038/s41375-021-01251-z
Mutational mechanisms shaping the coding and noncoding genome of germinal center derived B-cell lymphomas
Abstract
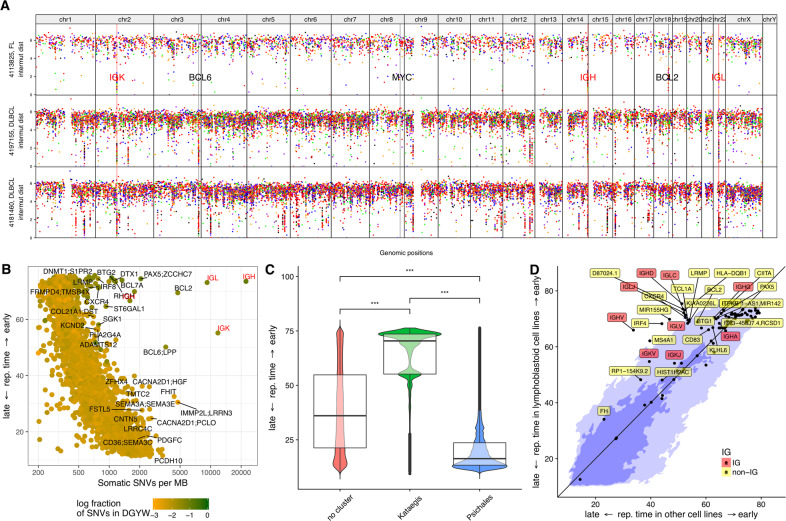
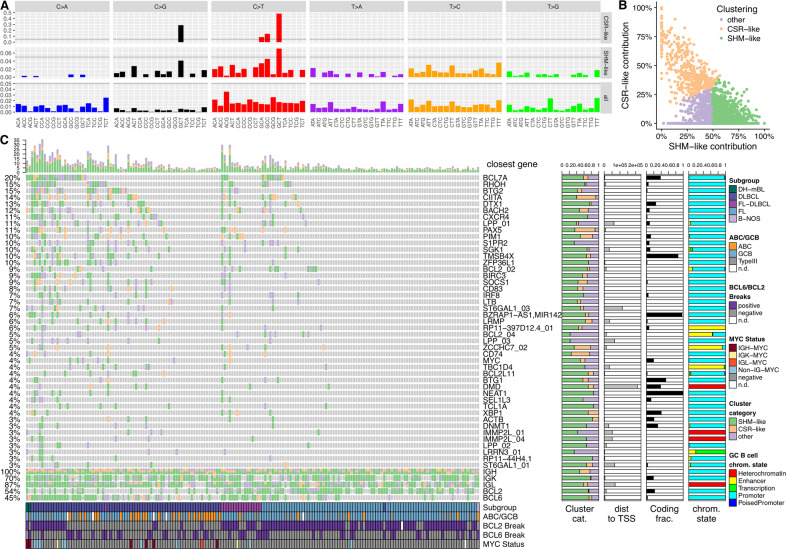
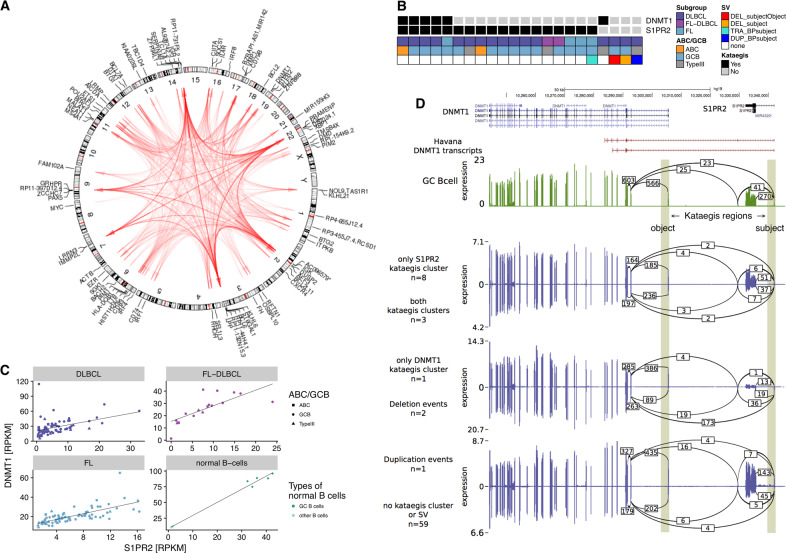
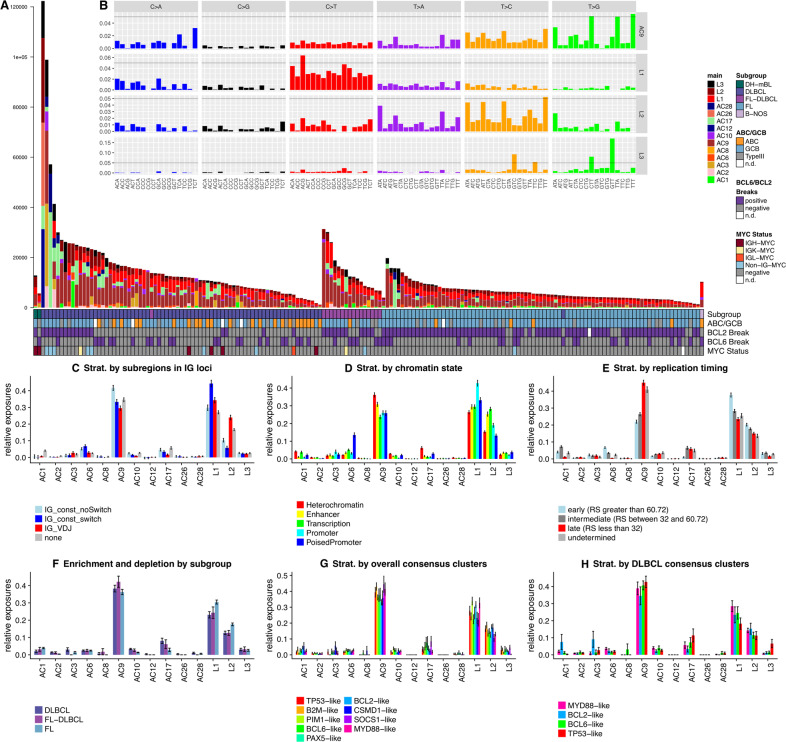
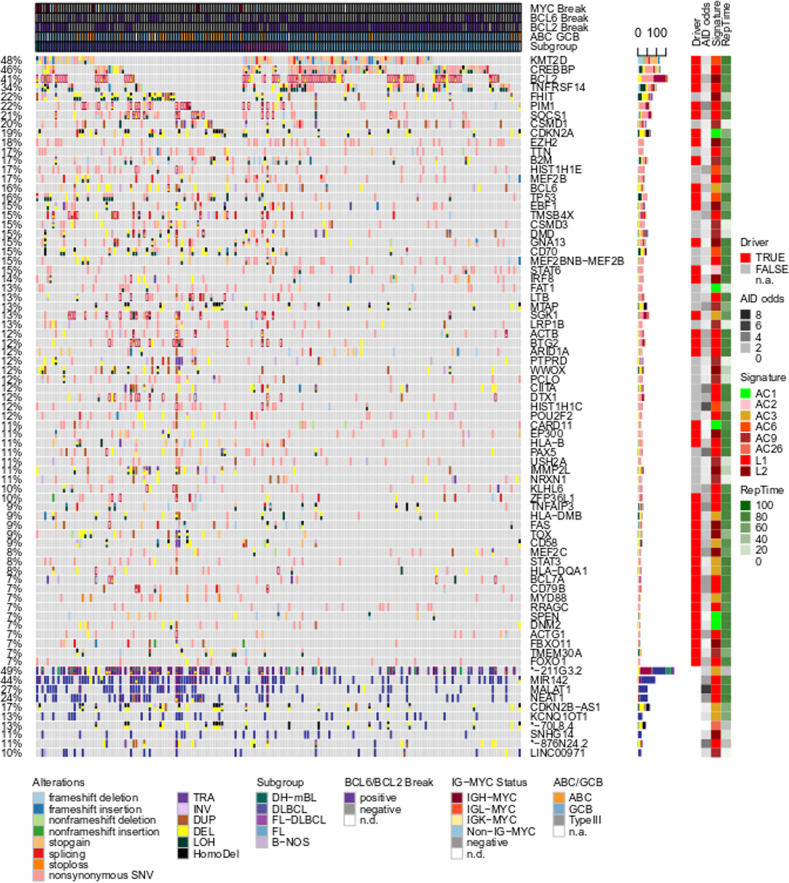
B cells have the unique property to somatically alter their immunoglobulin (IG) genes by V(D)J recombination, somatic hypermutation (SHM) and class-switch recombination (CSR). Aberrant targeting of these mechanisms is implicated in lymphomagenesis, but the mutational processes are poorly understood. By performing whole genome and transcriptome sequencing of 181 germinal center derived B-cell lymphomas (gcBCL) we identified distinct mutational signatures linked to SHM and CSR. We show that not only SHM, but presumably also CSR causes off-target mutations in non-IG genes. Kataegis clusters with high mutational density mainly affected early replicating regions and were enriched for SHM- and CSR-mediated off-target mutations. Moreover, they often co-occurred in loci physically interacting in the nucleus, suggesting that mutation hotspots promote increased mutation targeting of spatially co-localized loci (termed hypermutation by proxy). Only around 1% of somatic small variants were in protein coding sequences, but in about half of the driver genes, a contribution of B-cell specific mutational processes to their mutations was found. The B-cell-specific mutational processes contribute to both lymphoma initiation and intratumoral heterogeneity. Overall, we demonstrate that mutational processes involved in the development of gcBCL are more complex than previously appreciated, and that B cell-specific mutational processes contribute via diverse mechanisms to lymphomagenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Hummel M, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–30. - PubMed
-
- Rosenwald A, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
