

A high-quality bonobo genome refines the analysis of hominid evolution
- PMID: 33953399
- PMCID: PMC8172381
- DOI: 10.1038/s41586-021-03519-x
A high-quality bonobo genome refines the analysis of hominid evolution
Abstract
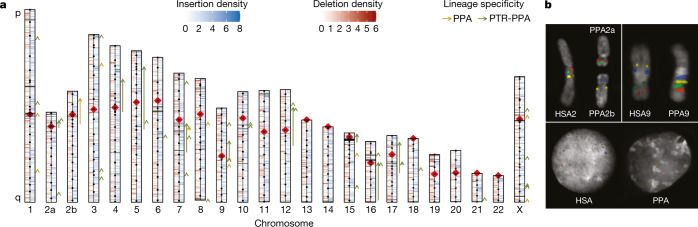
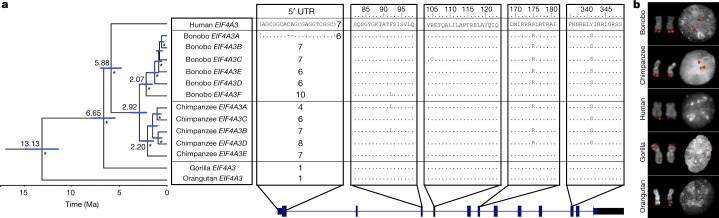
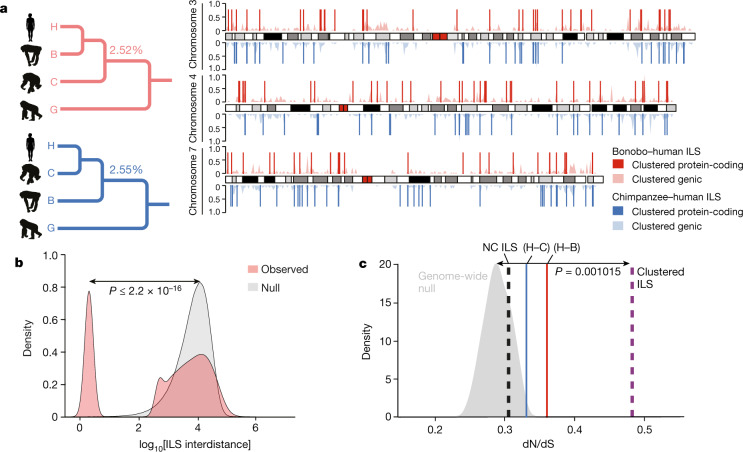
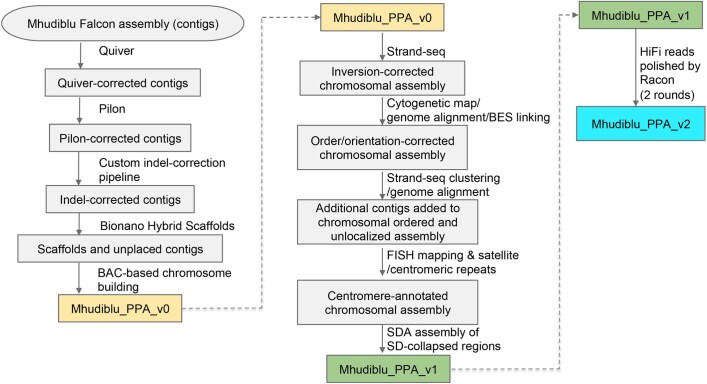
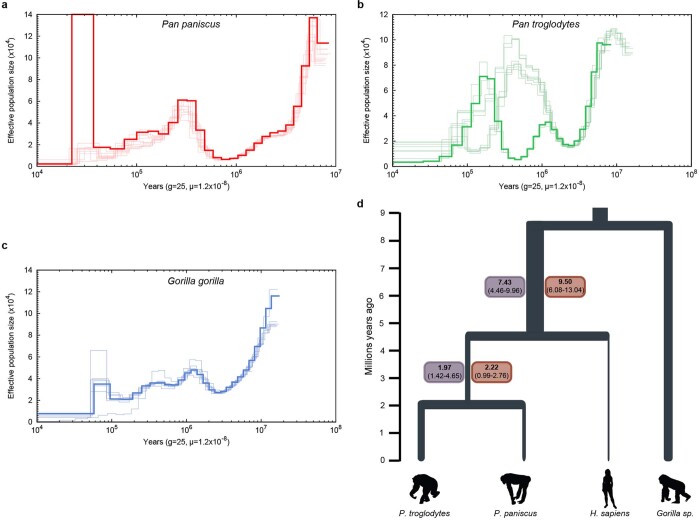
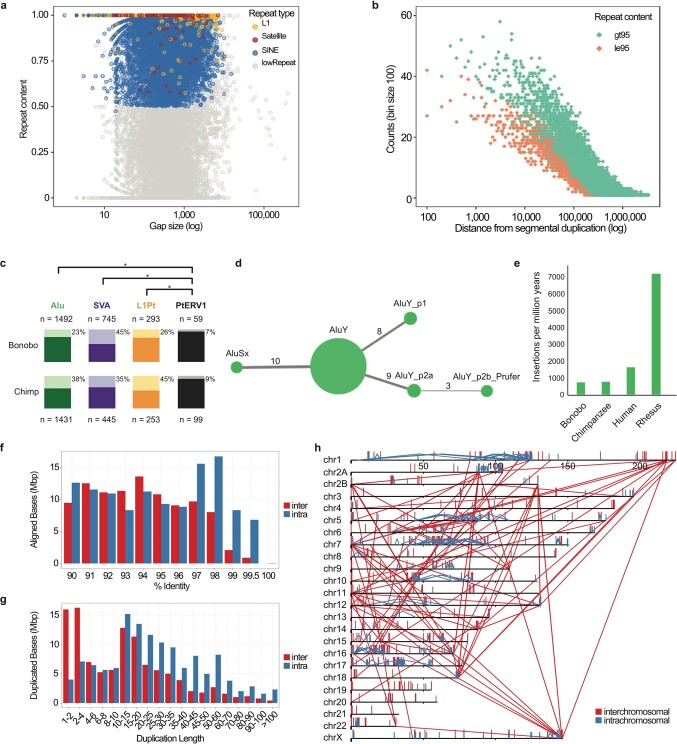
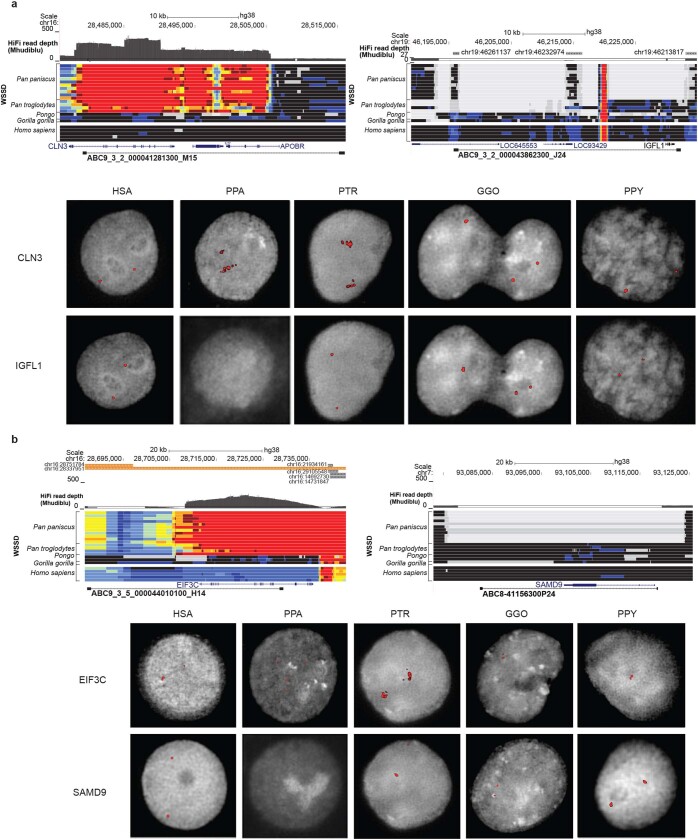
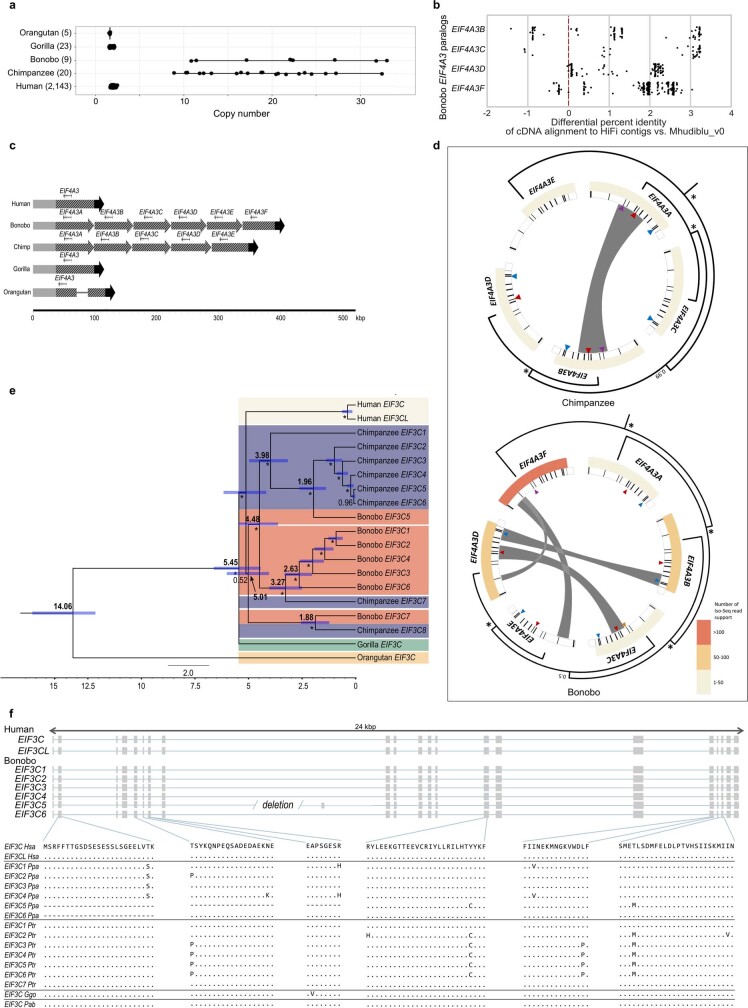
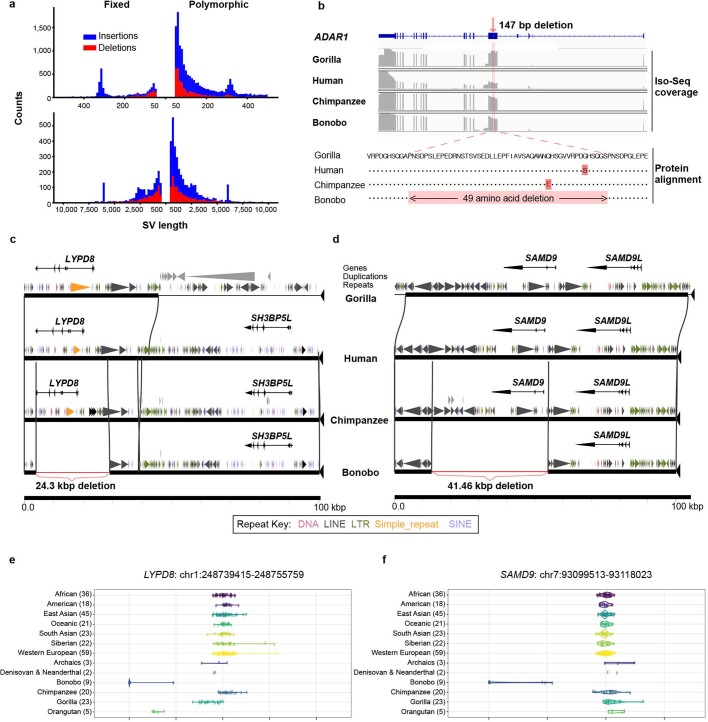
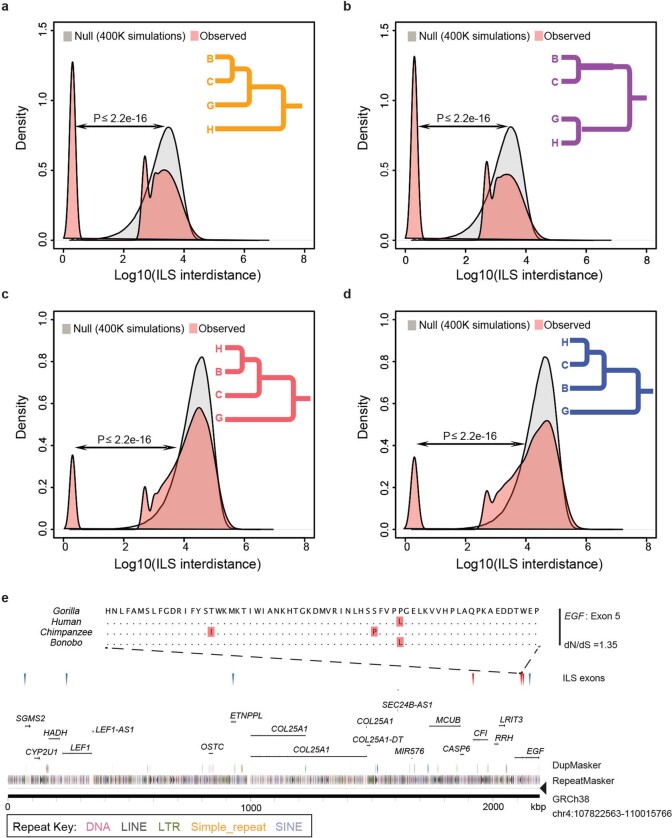
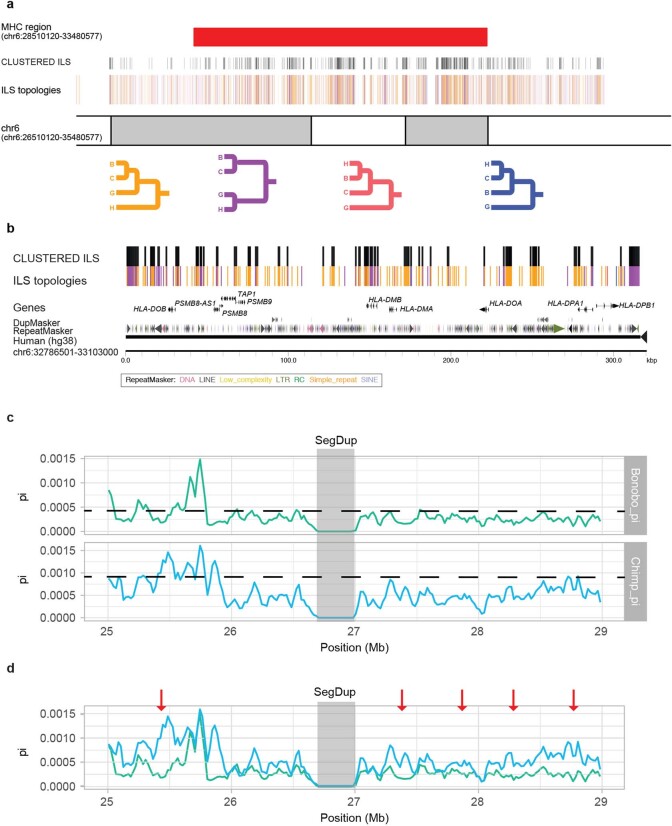
The divergence of chimpanzee and bonobo provides one of the few examples of recent hominid speciation1,2. Here we describe a fully annotated, high-quality bonobo genome assembly, which was constructed without guidance from reference genomes by applying a multiplatform genomics approach. We generate a bonobo genome assembly in which more than 98% of genes are completely annotated and 99% of the gaps are closed, including the resolution of about half of the segmental duplications and almost all of the full-length mobile elements. We compare the bonobo genome to those of other great apes1,3-5 and identify more than 5,569 fixed structural variants that specifically distinguish the bonobo and chimpanzee lineages. We focus on genes that have been lost, changed in structure or expanded in the last few million years of bonobo evolution. We produce a high-resolution map of incomplete lineage sorting and estimate that around 5.1% of the human genome is genetically closer to chimpanzee or bonobo and that more than 36.5% of the genome shows incomplete lineage sorting if we consider a deeper phylogeny including gorilla and orangutan. We also show that 26% of the segments of incomplete lineage sorting between human and chimpanzee or human and bonobo are non-randomly distributed and that genes within these clustered segments show significant excess of amino acid replacement compared to the rest of the genome.
Conflict of interest statement
J.G.U. is an employee of Pacific Biosciences. A.W.C.P., J.L. and A.R.H. are employees of Bionano Genomics. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- K99 HG011041/HG/NHGRI NIH HHS/United States
- U01 HG010961/HG/NHGRI NIH HHS/United States
- U01 HL137183/HL/NHLBI NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
- T32 HG008345/HG/NHGRI NIH HHS/United States
- R01 HG002939/HG/NHGRI NIH HHS/United States
- R01 GM059290/GM/NIGMS NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- U54 HG007990/HG/NHGRI NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
- T32 HG000035/HG/NHGRI NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- R01 HG010329/HG/NHGRI NIH HHS/United States
- U24 HG009081/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
