

Systematic interrogation of mutation groupings reveals divergent downstream expression programs within key cancer genes
- PMID: 33957863
- PMCID: PMC8101181
- DOI: 10.1186/s12859-021-04147-y
Systematic interrogation of mutation groupings reveals divergent downstream expression programs within key cancer genes
Abstract
Background: Genes implicated in tumorigenesis often exhibit diverse sets of genomic variants in the tumor cohorts within which they are frequently mutated. For many genes, neither the transcriptomic effects of these variants nor their relationship to one another in cancer processes have been well-characterized. We sought to identify the downstream expression effects of these mutations and to determine whether this heterogeneity at the genomic level is reflected in a corresponding heterogeneity at the transcriptomic level.
Results: By applying a novel hierarchical framework for organizing the mutations present in a cohort along with machine learning pipelines trained on samples' expression profiles we systematically interrogated the signatures associated with combinations of mutations recurrent in cancer. This allowed us to catalogue the mutations with discernible downstream expression effects across a number of tumor cohorts as well as to uncover and characterize over a hundred cases where subsets of a gene's mutations are clearly divergent in their function from the remaining mutations of the gene. These findings successfully replicated across a number of disease contexts and were found to have clear implications for the delineation of cancer processes and for clinical decisions.
Conclusions: The results of cataloguing the downstream effects of mutation subgroupings across cancer cohorts underline the importance of incorporating the diversity present within oncogenes in models designed to capture the downstream effects of their mutations.
Keywords: Cancer; Drug response; Genomic variants; Machine learning; Transcriptomics.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
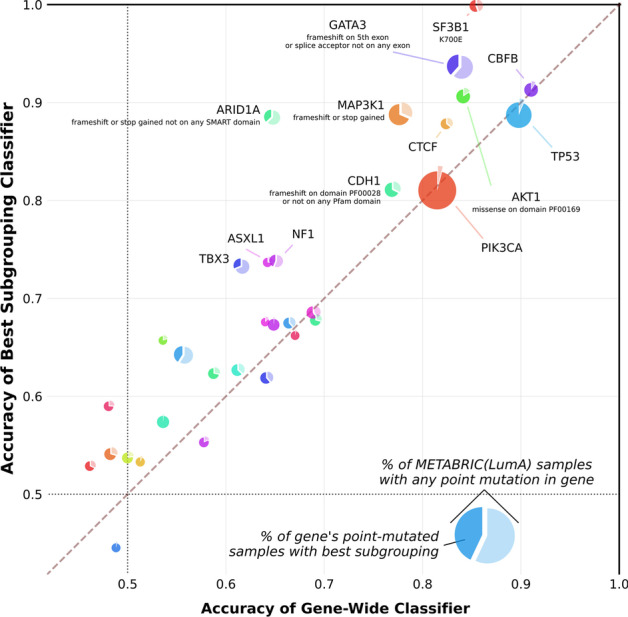
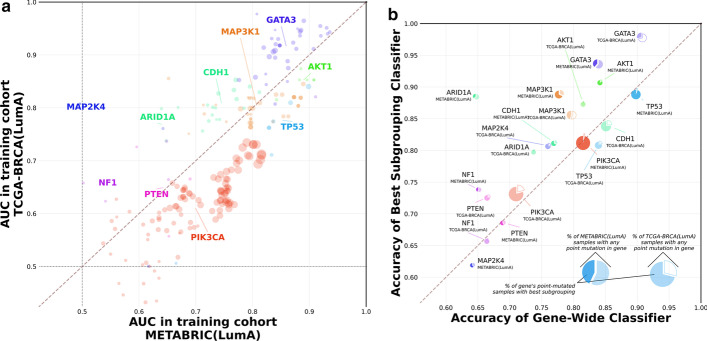
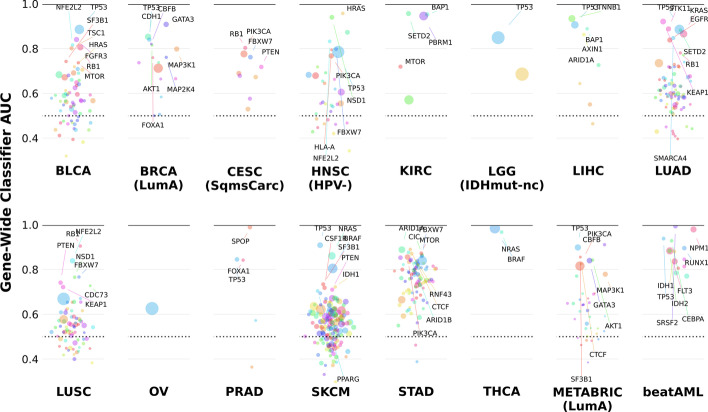
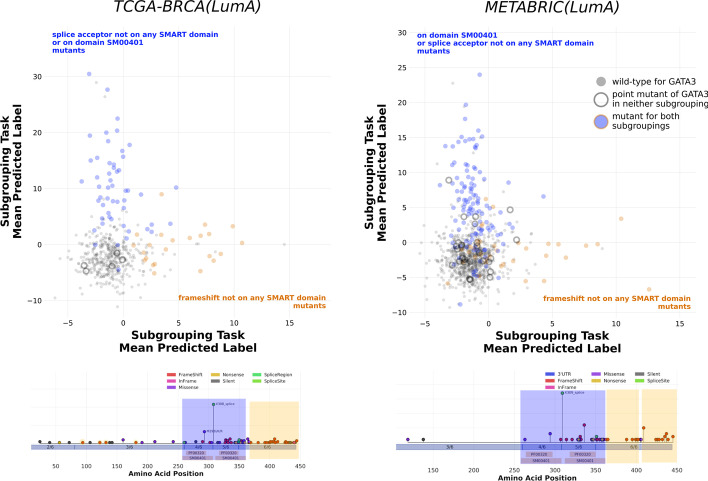
References
-
- Schram AM, Hyman DM. Quantifying the benefits of genome-driven oncology. Cancer Discov. 2017;7(6):552–4. doi: 10.1158/2159-8290.CD-17-0380. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
