

Improving deep learning-based protein distance prediction in CASP14
- PMID: 33961009
- PMCID: PMC8504632
- DOI: 10.1093/bioinformatics/btab355
Improving deep learning-based protein distance prediction in CASP14
Abstract
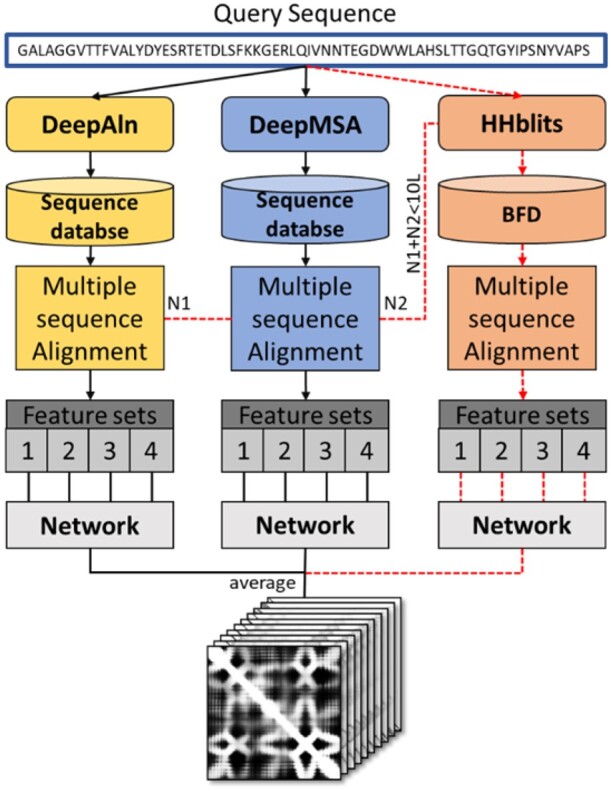
Motivation: Accurate prediction of residue-residue distances is important for protein structure prediction. We developed several protein distance predictors based on a deep learning distance prediction method and blindly tested them in the 14th Critical Assessment of Protein Structure Prediction (CASP14). The prediction method uses deep residual neural networks with the channel-wise attention mechanism to classify the distance between every two residues into multiple distance intervals. The input features for the deep learning method include co-evolutionary features as well as other sequence-based features derived from multiple sequence alignments (MSAs). Three alignment methods are used with multiple protein sequence/profile databases to generate MSAs for input feature generation. Based on different configurations and training strategies of the deep learning method, five MULTICOM distance predictors were created to participate in the CASP14 experiment.
Results: Benchmarked on 37 hard CASP14 domains, the best performing MULTICOM predictor is ranked 5th out of 30 automated CASP14 distance prediction servers in terms of precision of top L/5 long-range contact predictions [i.e. classifying distances between two residues into two categories: in contact (<8 Angstrom) and not in contact otherwise] and performs better than the best CASP13 distance prediction method. The best performing MULTICOM predictor is also ranked 6th among automated server predictors in classifying inter-residue distances into 10 distance intervals defined by CASP14 according to the precision of distance classification. The results show that the quality and depth of MSAs depend on alignment methods and sequence databases and have a significant impact on the accuracy of distance prediction. Using larger training datasets and multiple complementary features improves prediction accuracy. However, the number of effective sequences in MSAs is only a weak indicator of the quality of MSAs and the accuracy of predicted distance maps. In contrast, there is a strong correlation between the accuracy of contact/distance predictions and the average probability of the predicted contacts, which can therefore be more effectively used to estimate the confidence of distance predictions and select predicted distance maps.
Availability and implementation: The software package, source code and data of DeepDist2 are freely available at https://github.com/multicom-toolbox/deepdist and https://zenodo.org/record/4712084#.YIIM13VKhQM.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press.
Figures



Similar articles
-
Improving protein tertiary structure prediction by deep learning and distance prediction in CASP14.Proteins. 2022 Jan;90(1):58-72. doi: 10.1002/prot.26186. Epub 2021 Jul 27. Proteins. 2022. PMID: 34291486 Free PMC article.
-
MULTICOM2 open-source protein structure prediction system powered by deep learning and distance prediction.Sci Rep. 2021 Jun 23;11(1):13155. doi: 10.1038/s41598-021-92395-6. Sci Rep. 2021. PMID: 34162922 Free PMC article.
-
Analysis of several key factors influencing deep learning-based inter-residue contact prediction.Bioinformatics. 2020 Feb 15;36(4):1091-1098. doi: 10.1093/bioinformatics/btz679. Bioinformatics. 2020. PMID: 31504181 Free PMC article.
-
Recent Applications of Deep Learning Methods on Evolution- and Contact-Based Protein Structure Prediction.Int J Mol Sci. 2021 Jun 2;22(11):6032. doi: 10.3390/ijms22116032. Int J Mol Sci. 2021. PMID: 34199677 Free PMC article. Review.
-
Recent Advances in Protein Homology Detection Propelled by Inter-Residue Interaction Map Threading.Front Mol Biosci. 2021 May 11;8:643752. doi: 10.3389/fmolb.2021.643752. eCollection 2021. Front Mol Biosci. 2021. PMID: 34046429 Free PMC article. Review.
Cited by
-
Prediction of inter-chain distance maps of protein complexes with 2D attention-based deep neural networks.Nat Commun. 2022 Nov 15;13(1):6963. doi: 10.1038/s41467-022-34600-2. Nat Commun. 2022. PMID: 36379943 Free PMC article.
-
Freeprotmap: waiting-free prediction method for protein distance map.BMC Bioinformatics. 2024 May 4;25(1):176. doi: 10.1186/s12859-024-05771-0. BMC Bioinformatics. 2024. PMID: 38704533 Free PMC article.
-
Refinement of AlphaFold2 models against experimental and hybrid cryo-EM density maps.QRB Discov. 2022;3:e16. doi: 10.1017/qrd.2022.13. Epub 2022 Sep 20. QRB Discov. 2022. PMID: 37485023 Free PMC article.
-
High-Performance Deep Learning Toolbox for Genome-Scale Prediction of Protein Structure and Function.Workshop Mach Learn HPC Environ. 2021 Nov;2021:46-57. doi: 10.1109/mlhpc54614.2021.00010. Epub 2021 Dec 27. Workshop Mach Learn HPC Environ. 2021. PMID: 35112110 Free PMC article.
-
Improving protein tertiary structure prediction by deep learning and distance prediction in CASP14.Proteins. 2022 Jan;90(1):58-72. doi: 10.1002/prot.26186. Epub 2021 Jul 27. Proteins. 2022. PMID: 34291486 Free PMC article.
References
-
- Brünger A.T. et al. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr., 54, 905–921. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
