

Transcriptional and epi-transcriptional dynamics of SARS-CoV-2 during cellular infection
- PMID: 33961822
- PMCID: PMC8062406
- DOI: 10.1016/j.celrep.2021.109108
Transcriptional and epi-transcriptional dynamics of SARS-CoV-2 during cellular infection
Abstract
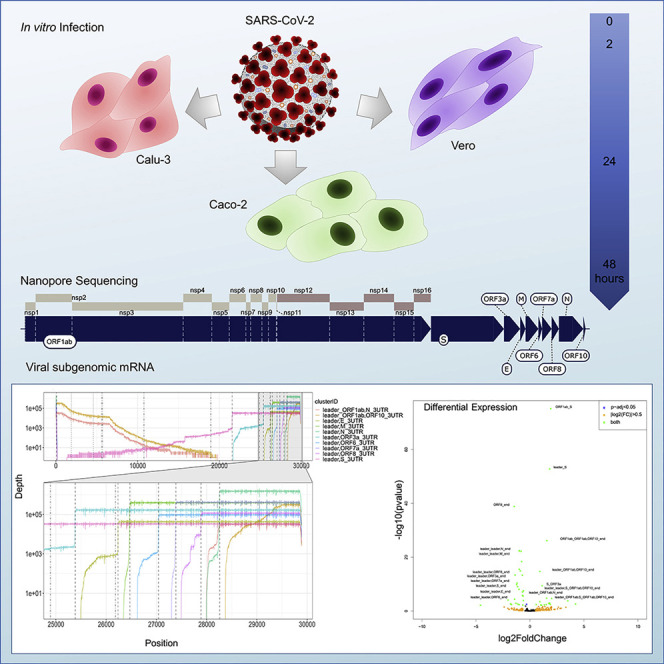
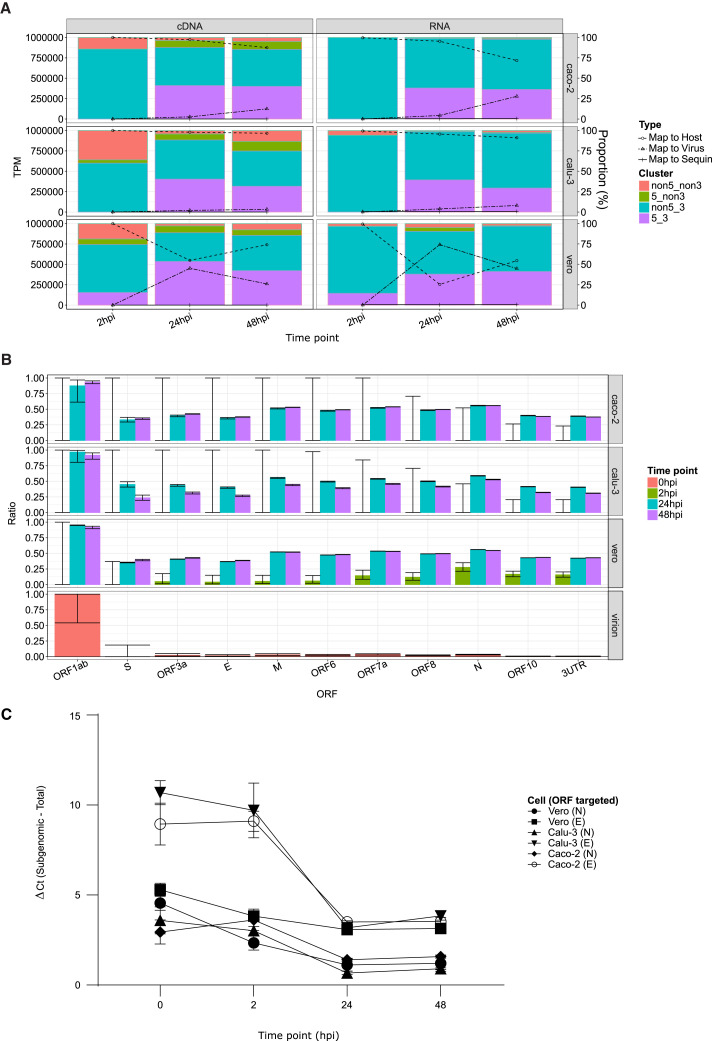
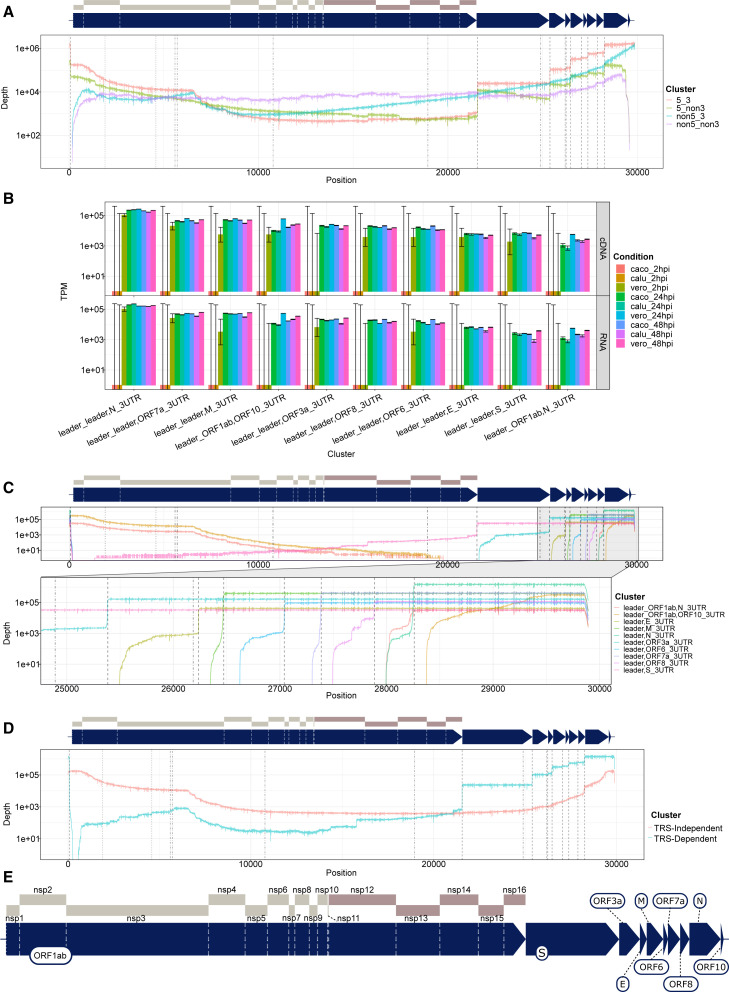
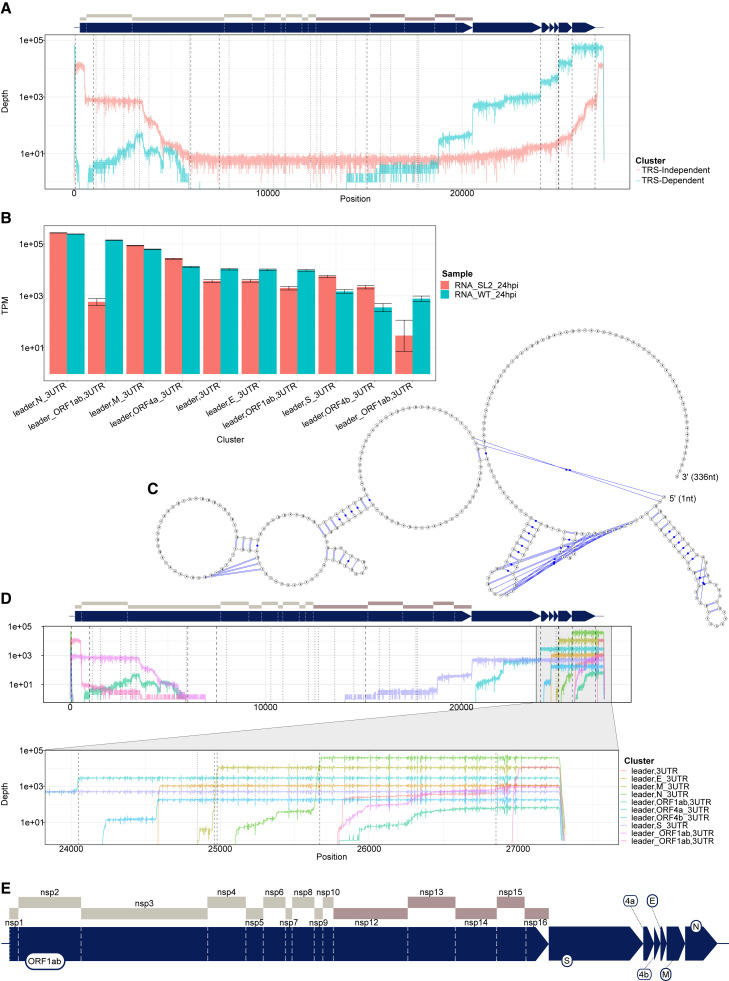
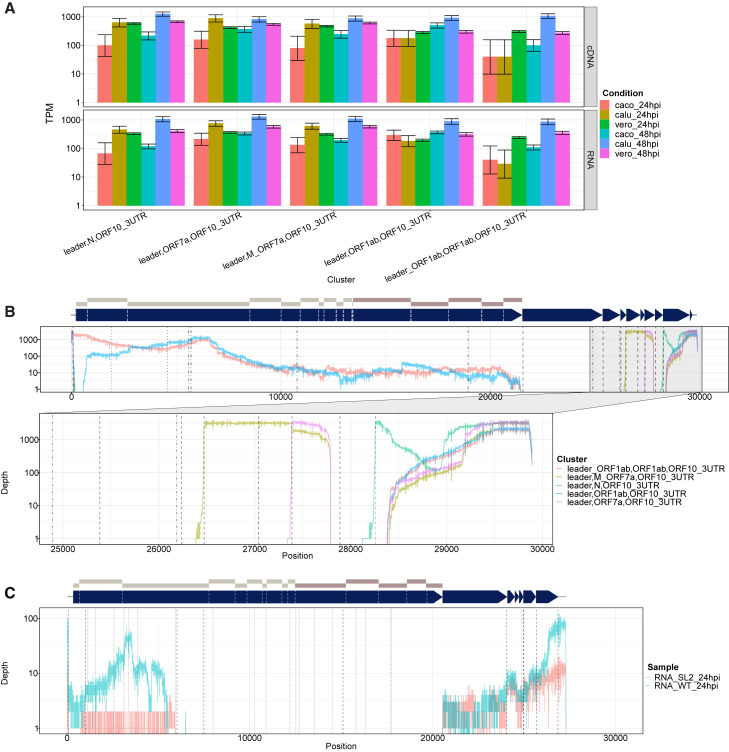
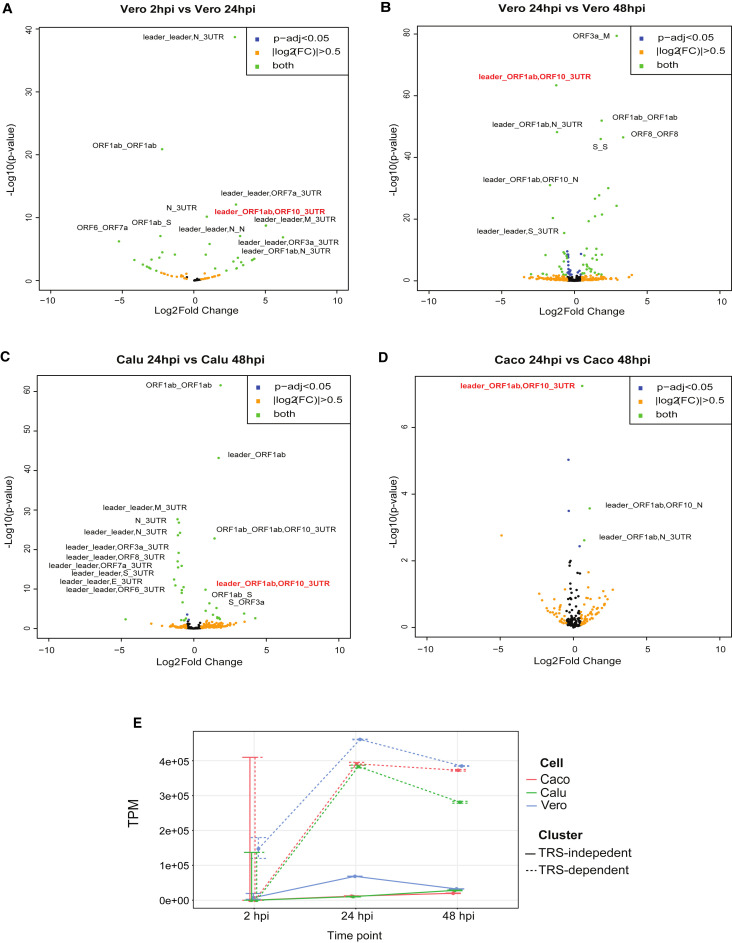
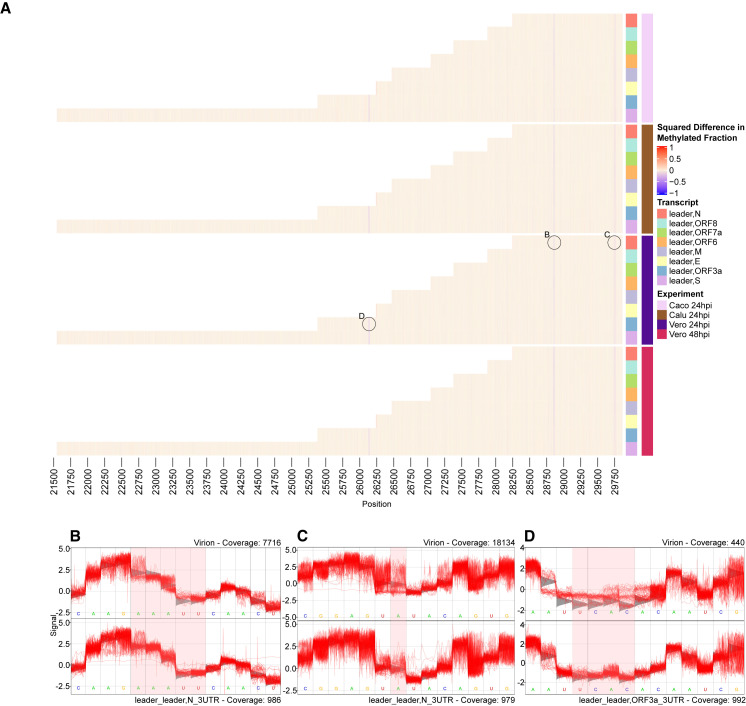
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) uses subgenomic RNA (sgRNA) to produce viral proteins for replication and immune evasion. We apply long-read RNA and cDNA sequencing to in vitro human and primate infection models to study transcriptional dynamics. Transcription-regulating sequence (TRS)-dependent sgRNA upregulates earlier in infection than TRS-independent sgRNA. An abundant class of TRS-independent sgRNA consisting of a portion of open reading frame 1ab (ORF1ab) containing nsp1 joins to ORF10, and the 3' untranslated region (UTR) upregulates at 48 h post-infection in human cell lines. We identify double-junction sgRNA containing both TRS-dependent and -independent junctions. We find multiple sites at which the SARS-CoV-2 genome is consistently more modified than sgRNA and that sgRNA modifications are stable across transcript clusters, host cells, and time since infection. Our work highlights the dynamic nature of the SARS-CoV-2 transcriptome during its replication cycle.
Keywords: COVID-19; Nanopore sequencing; RNA modification; SARS-CoV-2; coronavirus; differential expression; direct RNA sequencing; direct cDNA sequencing; discontinuous transcription; poly(A) tail.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests L.J.M.C., M.E.P., J.G., R.D.P.-I., and M.B.C. have received support from ONT to present their findings at scientific conferences. ONT played no role in study design, execution, analysis, or publication. L.J.M.C. has received research funding from ONT unrelated to this project.
Figures
References
-
- Davidson A.D., Williamson M.K., Lewis S., Shoemark D., Carroll M.W., Heesom K.J., Zambon M., Ellis J., Lewis P.A., Hiscox J.A., Matthews D.A. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020;12:68. - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
