

Adenosine kinase: An epigenetic modulator in development and disease
- PMID: 33961946
- PMCID: PMC8178237
- DOI: 10.1016/j.neuint.2021.105054
Adenosine kinase: An epigenetic modulator in development and disease
Abstract
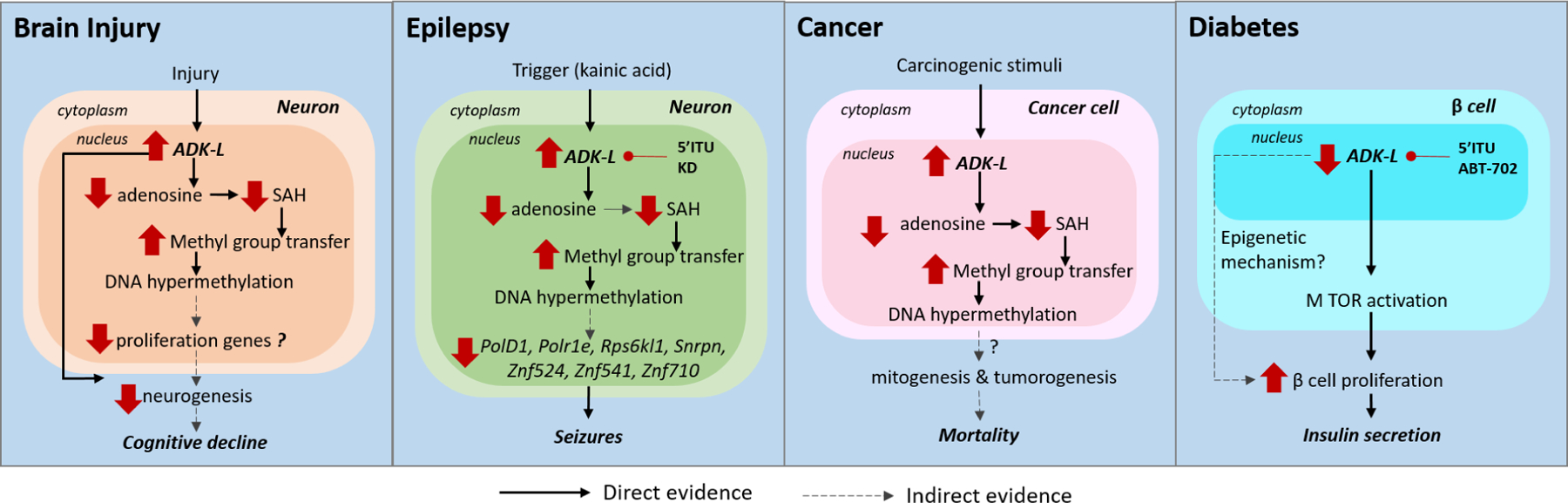
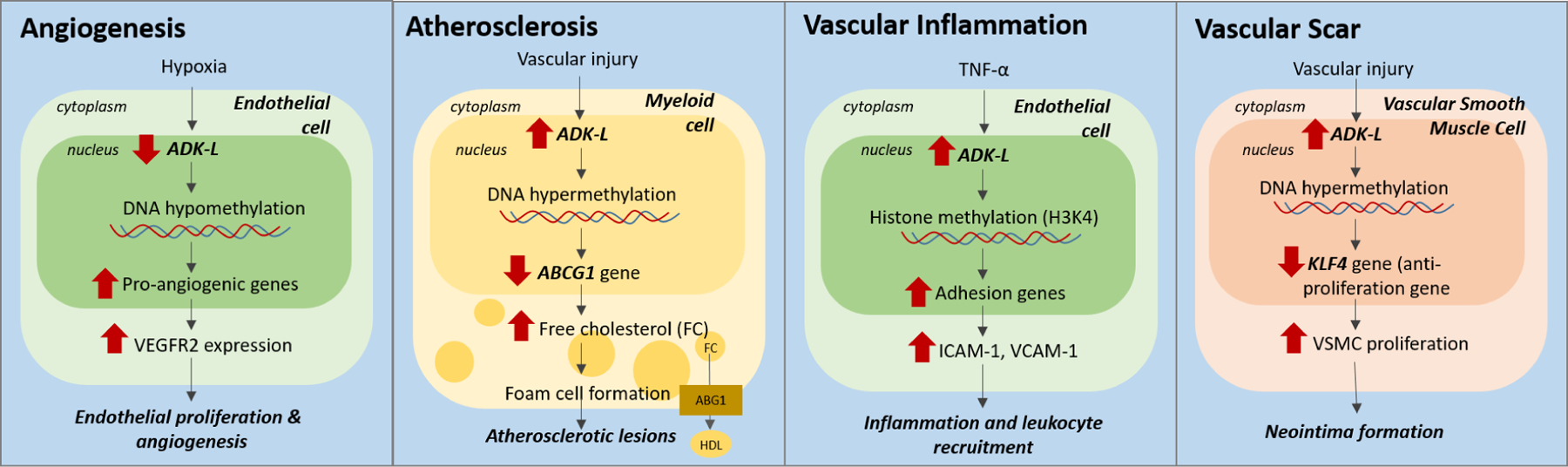
Adenosine kinase (ADK) is the key regulator of adenosine and catalyzes the metabolism of adenosine to 5'-adenosine monophosphate. The enzyme exists in two isoforms: a long isoform (ADK-long, ADK-L) and a short isoform (ADK-short, ADK-S). The two isoforms are developmentally regulated and are differentially expressed in distinct subcellular compartments with ADK-L localized in the nucleus and ADK-S localized in the cytoplasm. The nuclear localization of ADK-L and its biochemical link to the transmethylation pathway suggest a specific role for gene regulation via epigenetic mechanisms. Recent evidence reveals an adenosine receptor-independent role of ADK in determining the global methylation status of DNA and thereby contributing to epigenomic regulation. Here we summarize recent progress in understanding the biochemical interactions between adenosine metabolism by ADK-L and epigenetic modifications linked to transmethylation reactions. This review will provide a comprehensive overview of ADK-associated changes in DNA methylation in developmental, as well as in pathological conditions including brain injury, epilepsy, vascular diseases, cancer, and diabetes. Challenges in investigating the epigenetic role of ADK for therapeutic gains are briefly discussed.
Keywords: Adenosine kinase; DNA methylation; Development; Epigenetic regulator; Epilepsy.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests
⊠The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures


Similar articles
-
Adenosine Kinase: An Epigenetic Modulator and Drug Target.J Inherit Metab Dis. 2025 May;48(3):e70033. doi: 10.1002/jimd.70033. J Inherit Metab Dis. 2025. PMID: 40393929 Free PMC article. Review.
-
The role of adenosine in epilepsy.Brain Res Bull. 2019 Sep;151:46-54. doi: 10.1016/j.brainresbull.2018.11.008. Epub 2018 Nov 20. Brain Res Bull. 2019. PMID: 30468847 Free PMC article. Review.
-
Adenosine kinase: exploitation for therapeutic gain.Pharmacol Rev. 2013 Apr 16;65(3):906-43. doi: 10.1124/pr.112.006361. Print 2013 Jul. Pharmacol Rev. 2013. PMID: 23592612 Free PMC article. Review.
-
Adenosine Kinase: Cytoplasmic and Nuclear Isoforms.In: Noebels JL, Avoli M, Rogawski MA, Vezzani A, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. 5th edition. New York: Oxford University Press; 2024. Chapter 27. In: Noebels JL, Avoli M, Rogawski MA, Vezzani A, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. 5th edition. New York: Oxford University Press; 2024. Chapter 27. PMID: 39637220 Free Books & Documents. Review.
-
Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus.Biochem Biophys Res Commun. 2009 Oct 9;388(1):46-50. doi: 10.1016/j.bbrc.2009.07.106. Epub 2009 Jul 25. Biochem Biophys Res Commun. 2009. PMID: 19635462
Cited by
-
Ectonucleotidases as bridge between the ATP and adenosine world: reflections on Geoffrey Burnstock.Purinergic Signal. 2022 Jun;18(2):193-198. doi: 10.1007/s11302-022-09862-6. Epub 2022 May 6. Purinergic Signal. 2022. PMID: 35522386 Free PMC article. Review.
-
Brain Proteome-Wide Association Study Identifies Candidate Genes that Regulate Protein Abundance Associated with Post-Traumatic Stress Disorder.Genes (Basel). 2022 Jul 27;13(8):1341. doi: 10.3390/genes13081341. Genes (Basel). 2022. PMID: 35893077 Free PMC article.
-
Emerging roles of dysregulated adenosine homeostasis in brain disorders with a specific focus on neurodegenerative diseases.J Biomed Sci. 2021 Oct 11;28(1):70. doi: 10.1186/s12929-021-00766-y. J Biomed Sci. 2021. PMID: 34635103 Free PMC article. Review.
-
Deciphering adenosine signaling in hepatocellular carcinoma: Pathways, prognostic models, and therapeutic implications.Clin Mol Hepatol. 2025 Jul;31(3):706-729. doi: 10.3350/cmh.2024.1068. Epub 2025 Feb 5. Clin Mol Hepatol. 2025. PMID: 39905839 Free PMC article. Review.
-
Hepatocyte Adenosine Kinase Promotes Excessive Fat Deposition and Liver Inflammation.Gastroenterology. 2023 Jan;164(1):134-146. doi: 10.1053/j.gastro.2022.09.027. Epub 2022 Sep 28. Gastroenterology. 2023. PMID: 36181835 Free PMC article.
References
-
- Ahmed Abdalhamid Osman M, Sun YJ, Li RJ, Lin H, Zeng DM, Chen XY, He D, Feng HW, Yang Z, Wang J, Wu C, Cui M, Sun JP, Huo Y, Yu X, 2019. Deletion of pancreatic beta-cell adenosine kinase improves glucose homeostasis in young mice and ameliorates streptozotocin-induced hyperglycaemia. J Cell Mol Med 23, 4653–4665. - PMC - PubMed
-
- Amisten S, Braun OO, Bengtsson A, Erlinge D, 2008. Gene expression profiling for the identification of G-protein coupled receptors in human platelets. Thromb Res 122, 47–57. - PubMed
-
- Annegers JF, Coan SP, 2000. The risks of epilepsy after traumatic brain injury. Seizure 9, 453–457. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
