

Excitation spectral microscopy for highly multiplexed fluorescence imaging and quantitative biosensing
- PMID: 33963178
- PMCID: PMC8105378
- DOI: 10.1038/s41377-021-00536-3
Excitation spectral microscopy for highly multiplexed fluorescence imaging and quantitative biosensing
Abstract
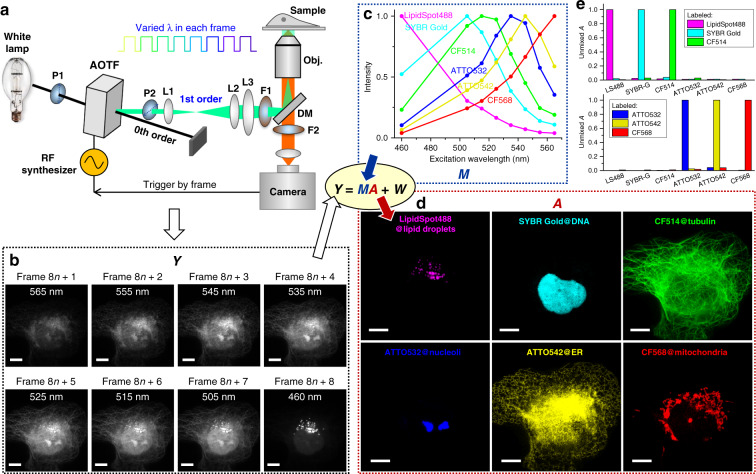
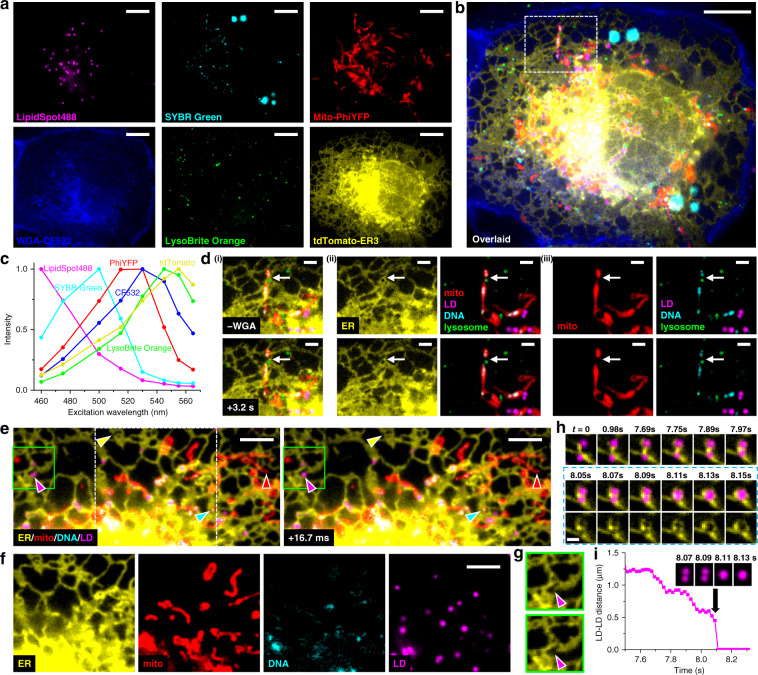
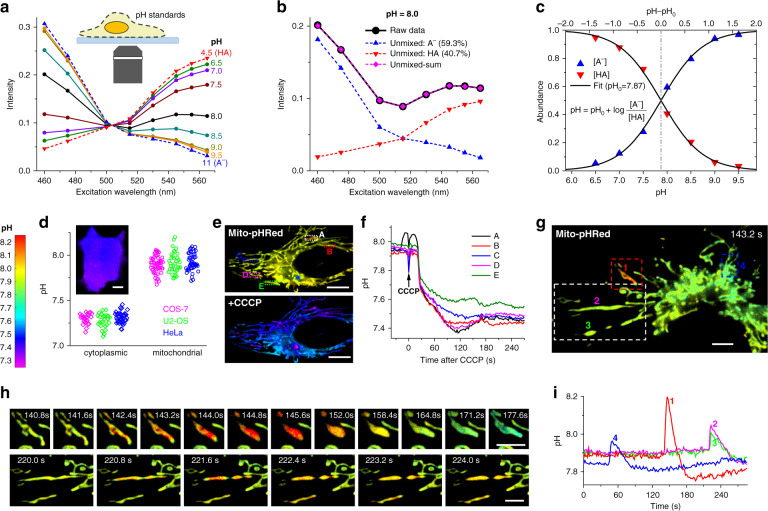
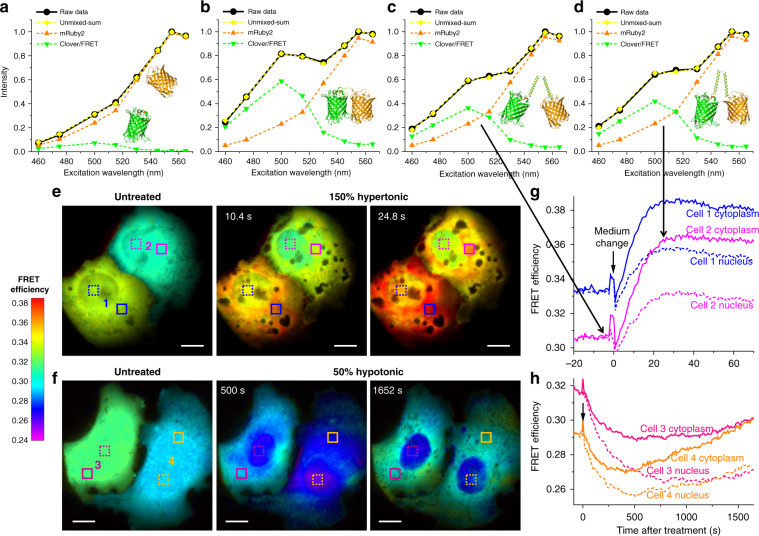
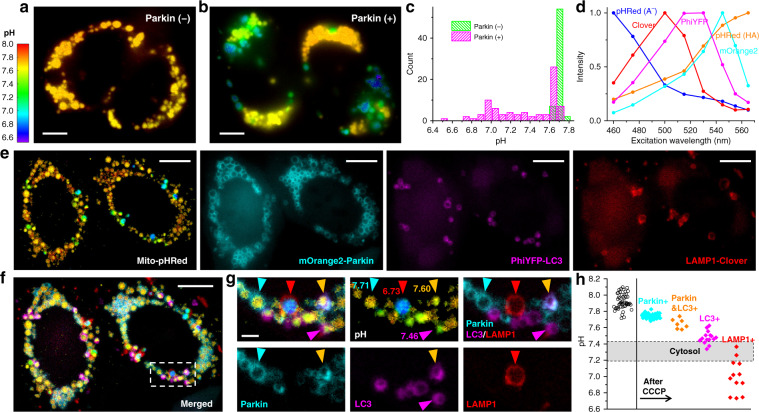
The multiplexing capability of fluorescence microscopy is severely limited by the broad fluorescence spectral width. Spectral imaging offers potential solutions, yet typical approaches to disperse the local emission spectra notably impede the attainable throughput. Here we show that using a single, fixed fluorescence emission detection band, through frame-synchronized fast scanning of the excitation wavelength from a white lamp via an acousto-optic tunable filter, up to six subcellular targets, labeled by common fluorophores of substantial spectral overlap, can be simultaneously imaged in live cells with low (~1%) crosstalks and high temporal resolutions (down to ~10 ms). The demonstrated capability to quantify the abundances of different fluorophores in the same sample through unmixing the excitation spectra next enables us to devise novel, quantitative imaging schemes for both bi-state and Förster resonance energy transfer fluorescent biosensors in live cells. We thus achieve high sensitivities and spatiotemporal resolutions in quantifying the mitochondrial matrix pH and intracellular macromolecular crowding, and further demonstrate, for the first time, the multiplexing of absolute pH imaging with three additional target organelles/proteins to elucidate the complex, Parkin-mediated mitophagy pathway. Together, excitation spectral microscopy provides exceptional opportunities for highly multiplexed fluorescence imaging. The prospect of acquiring fast spectral images without the need for fluorescence dispersion or care for the spectral response of the detector offers tremendous potential.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Super-multiplexing excitation spectral microscopy with multiple fluorescence bands.Biomed Opt Express. 2022 Oct 27;13(11):6048-6060. doi: 10.1364/BOE.473241. eCollection 2022 Nov 1. Biomed Opt Express. 2022. PMID: 36733753 Free PMC article.
-
Spectrally Resolved and Functional Super-resolution Microscopy via Ultrahigh-Throughput Single-Molecule Spectroscopy.Acc Chem Res. 2018 Mar 20;51(3):697-705. doi: 10.1021/acs.accounts.7b00545. Epub 2018 Feb 14. Acc Chem Res. 2018. PMID: 29443498
-
Multiplexed imaging in live cells using pulsed interleaved excitation spectral FLIM.Opt Express. 2024 Jan 29;32(3):3290-3307. doi: 10.1364/OE.505667. Opt Express. 2024. PMID: 38297554 Free PMC article.
-
Live-cell fluorescence spectral imaging as a data science challenge.Biophys Rev. 2022 Mar 23;14(2):579-597. doi: 10.1007/s12551-022-00941-x. eCollection 2022 Apr. Biophys Rev. 2022. PMID: 35528031 Free PMC article. Review.
-
Spectral imaging and its applications in live cell microscopy.FEBS Lett. 2003 Jul 3;546(1):87-92. doi: 10.1016/s0014-5793(03)00521-0. FEBS Lett. 2003. PMID: 12829241 Review.
Cited by
-
Highly Efficient Calibration-Free Color Compensation Algorithm for Imaging Flow Cytometry.Cytometry A. 2025 May;107(5):309-320. doi: 10.1002/cyto.a.24931. Epub 2025 Apr 9. Cytometry A. 2025. PMID: 40202100
-
AutoUnmix: an autoencoder-based spectral unmixing method for multi-color fluorescence microscopy imaging.Biomed Opt Express. 2023 Aug 22;14(9):4814-4827. doi: 10.1364/BOE.498421. eCollection 2023 Sep 1. Biomed Opt Express. 2023. PMID: 37791286 Free PMC article.
-
Droplet microfluidics for single-cell studies: a frontier in ecological understanding of microbiomes.FEMS Microbiol Rev. 2025 Jan 14;49:fuaf032. doi: 10.1093/femsre/fuaf032. FEMS Microbiol Rev. 2025. PMID: 40699006 Free PMC article. Review.
-
High-speed fluorescence excitation-scanning hyperspectral imaging microscopy using thin-film tunable filters.Proc SPIE Int Soc Opt Eng. 2024 Jan-Feb;12846:128460B. doi: 10.1117/12.3001600. Epub 2024 Mar 12. Proc SPIE Int Soc Opt Eng. 2024. PMID: 38577224 Free PMC article.
-
Super-multiplex imaging of cellular dynamics and heterogeneity by integrated stimulated Raman and fluorescence microscopy.iScience. 2021 Jul 9;24(8):102832. doi: 10.1016/j.isci.2021.102832. eCollection 2021 Aug 20. iScience. 2021. PMID: 34381966 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
