

LZerD webserver for pairwise and multiple protein-protein docking
- PMID: 33963854
- PMCID: PMC8262708
- DOI: 10.1093/nar/gkab336
LZerD webserver for pairwise and multiple protein-protein docking
Abstract
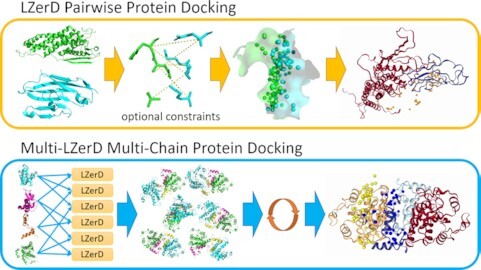
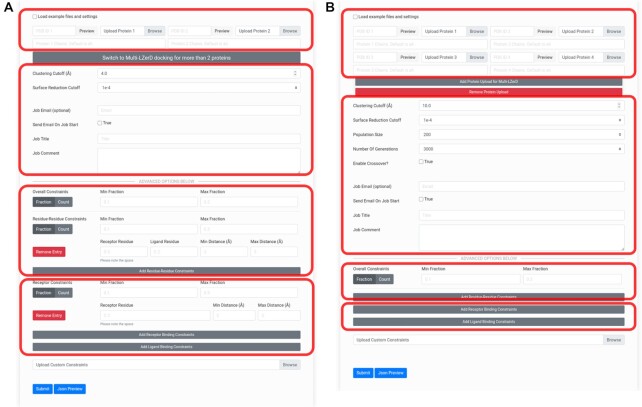
Protein complexes are involved in many important processes in living cells. To understand the mechanisms of these processes, it is necessary to solve the 3D structures of the protein complexes. When protein complex structures have not yet been determined by experiment, protein-protein docking tools can be used to computationally model the structures of these complexes. Here, we present a webserver which provides access to LZerD and Multi-LZerD protein docking tools. The protocol provided by the server have performed consistently among the top in the CAPRI blind evaluation. LZerD docks pairs of structures, while Multi-LZerD can dock three or more structures simultaneously. LZerD uses a soft protein surface representation with 3D Zernike descriptors and explores the binding pose space using geometric hashing. Multi-LZerD performs multi-chain docking by combining pairwise solutions by LZerD. Both methods output full-atom docked models of the input proteins. Users can also input distance constraints between interacting or non-interacting residues as well as residues that locate at the interface or far from the interface. The webserver is equipped with a user-friendly panel that visualizes the distribution and structures of binding poses of top scoring models. The LZerD webserver is available at https://lzerd.kiharalab.org.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
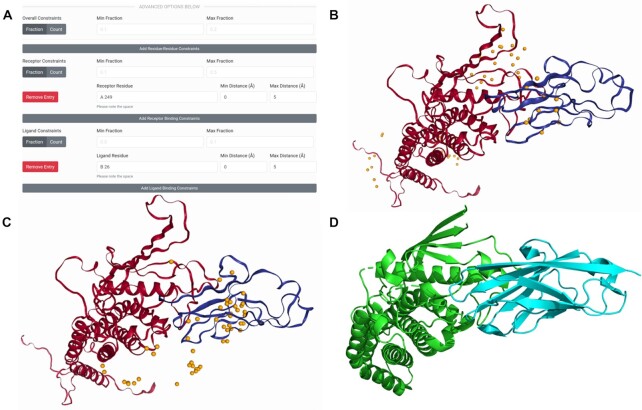
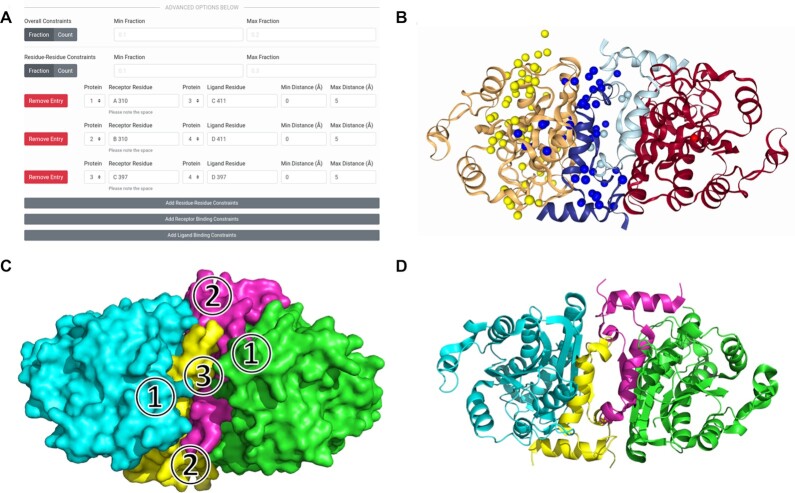
 has an I-RMSD of 1.05 Å and an
has an I-RMSD of 1.05 Å and an  has an I-RMSD of 1.38 Å and an
has an I-RMSD of 1.38 Å and an  has an I-RMSD of 1.02 and an
has an I-RMSD of 1.02 and an References
-
- Mintseris J., Pierce B., Wiehe K., Anderson R., Chen R., Weng Z.. Integrating statistical pair potentials into protein complex prediction. Proteins. 2007; 69:511–520. - PubMed
-
- Dominguez C., Boelens R., Bonvin A.M.. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003; 125:1731–1737. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
