

Nephroplex: a kidney-focused NGS panel highlights the challenges of PKD1 sequencing and identifies a founder BBS4 mutation
- PMID: 33964006
- PMCID: PMC8610957
- DOI: 10.1007/s40620-021-01048-4
Nephroplex: a kidney-focused NGS panel highlights the challenges of PKD1 sequencing and identifies a founder BBS4 mutation
Abstract
Background: Genetic testing of patients with inherited kidney diseases has emerged as a tool of clinical utility by improving the patients' diagnosis, prognosis, surveillance and therapy.
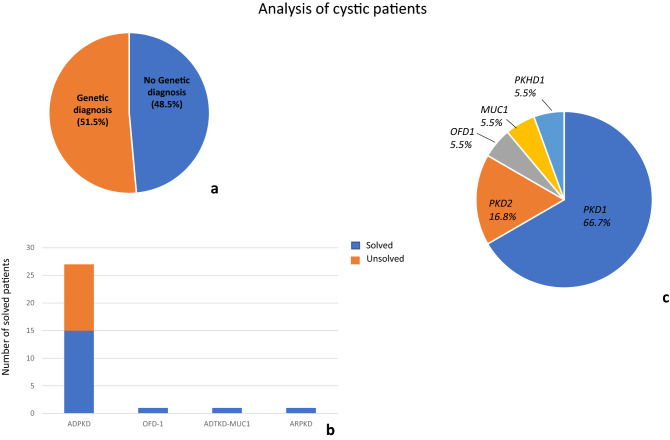
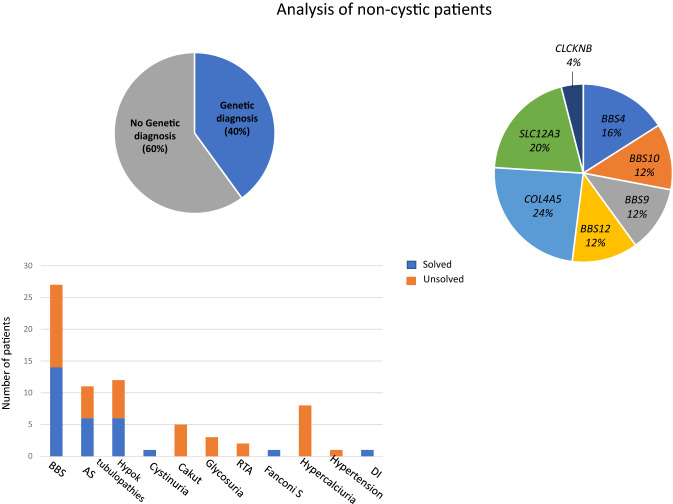
Methods: The present study applied a Next Generation Sequencing (NGS)-based panel, named NephroPlex, testing 115 genes causing renal diseases, to 119 individuals, including 107 probands and 12 relatives. Thirty-five (poly)cystic and 72 non (poly)cystic individuals were enrolled. The latter subgroup of patients included Bardet-Biedl syndrome (BBS) patients, as major components.
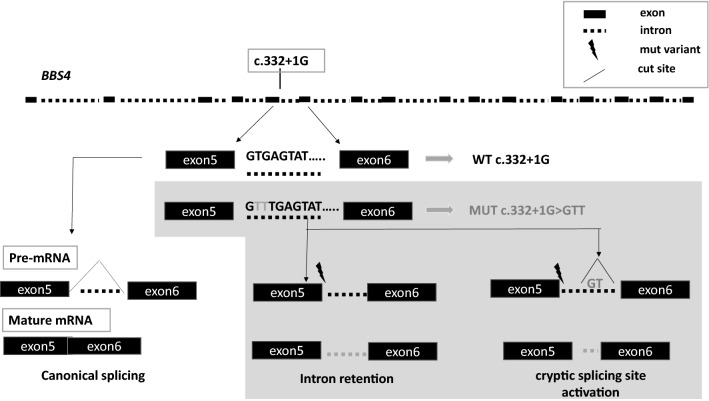
Results: Disease-causing mutations were identified in 51.5 and 40% of polycystic and non-polycystic individuals, respectively. Autosomal dominant polycystic kidney disease (ADPKD) patients with truncating PKD1 variants showed a trend towards a greater slope of the age-estimated glomerular filtration rate (eGFR) regression line than patients with (i) missense variants, (ii) any PKD2 mutations and (iii) no detected mutations, according to previous findings. The analysis of BBS individuals showed a similar frequency of BBS4,9,10 and 12 mutations. Of note, all BBS4-mutated patients harbored the novel c.332+1G>GTT variant, which was absent in public databases, however, in our internal database, an additional heterozygote carrier was found. All BBS4-mutated individuals originated from the same geographical area encompassing the coastal provinces of Naples.
Discussion: In conclusion, these findings indicate the potential for a genetic panel to provide useful information at both clinical and epidemiological levels.
Keywords: ADPKD; Bardet-Biedl syndrome; Gene-panel; Inherited kidney disease; NGS.
© 2021. The Author(s).
Conflict of interest statement
The authors have no conflict of interest to declare.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
