

A mutation in SLC37A4 causes a dominantly inherited congenital disorder of glycosylation characterized by liver dysfunction
- PMID: 33964207
- PMCID: PMC8206404
- DOI: 10.1016/j.ajhg.2021.04.013
A mutation in SLC37A4 causes a dominantly inherited congenital disorder of glycosylation characterized by liver dysfunction
Abstract
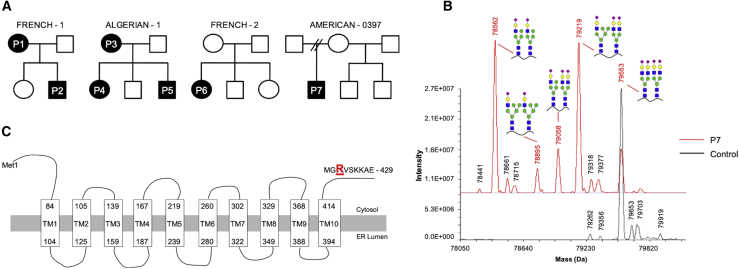
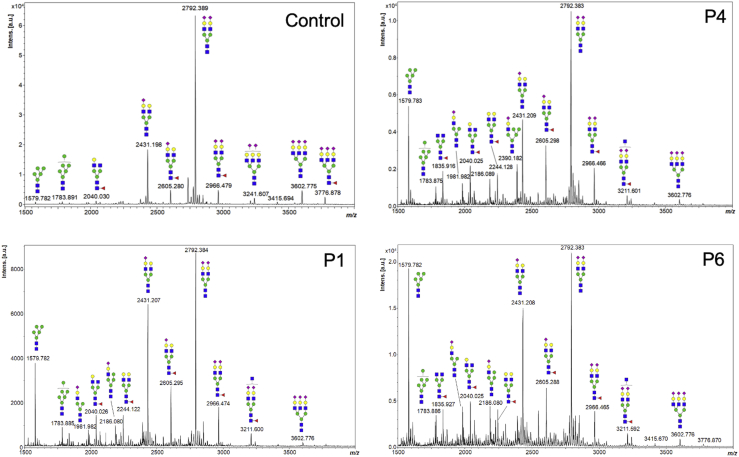
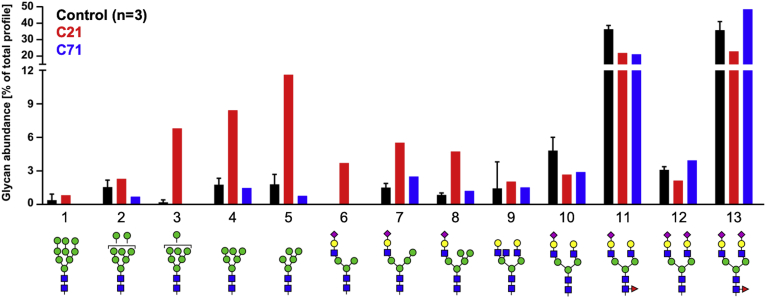
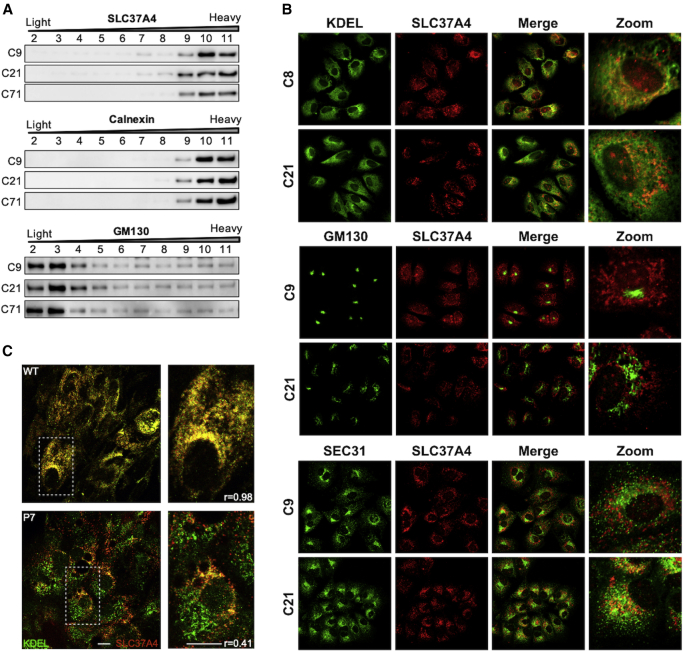
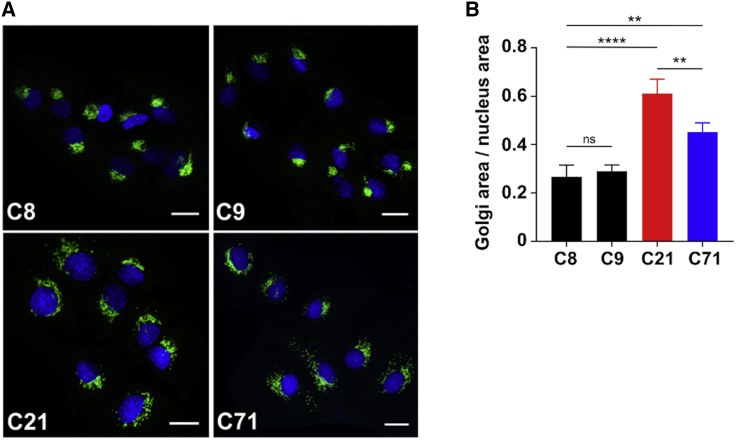
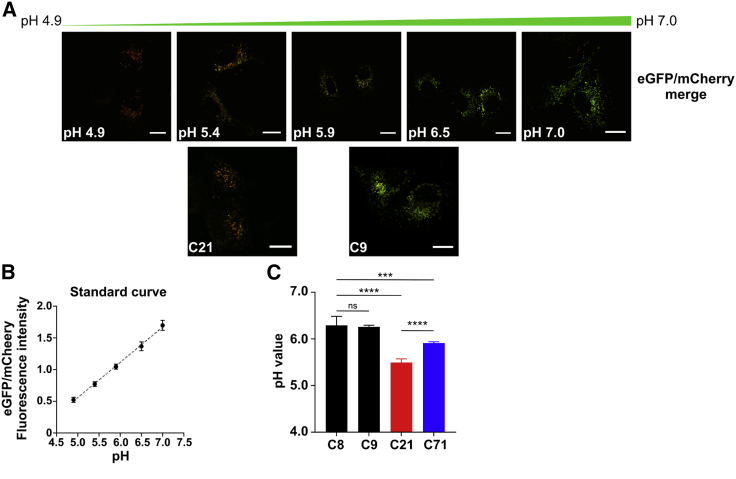
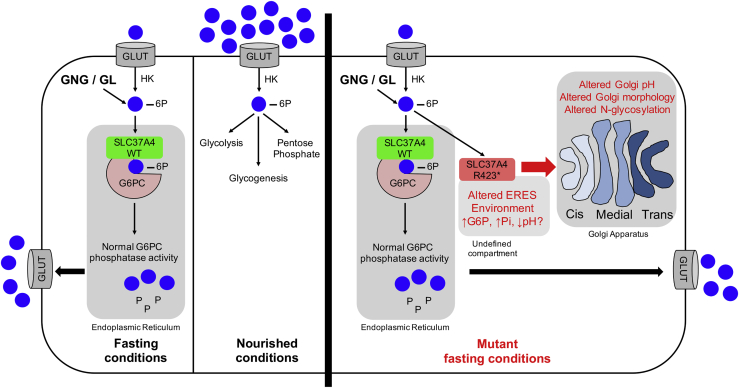
SLC37A4 encodes an endoplasmic reticulum (ER)-localized multitransmembrane protein required for transporting glucose-6-phosphate (Glc-6P) into the ER. Once transported into the ER, Glc-6P is subsequently hydrolyzed by tissue-specific phosphatases to glucose and inorganic phosphate during times of glucose depletion. Pathogenic variants in SLC37A4 cause an established recessive disorder known as glycogen storage disorder 1b characterized by liver and kidney dysfunction with neutropenia. We report seven individuals who presented with liver dysfunction multifactorial coagulation deficiency and cardiac issues and were heterozygous for the same variant, c.1267C>T (p.Arg423∗), in SLC37A4; the affected individuals were from four unrelated families. Serum samples from affected individuals showed profound accumulation of both high mannose and hybrid type N-glycans, while N-glycans in fibroblasts and undifferentiated iPSC were normal. Due to the liver-specific nature of this disorder, we generated a CRISPR base-edited hepatoma cell line harboring the c.1267C>T (p.Arg423∗) variant. These cells replicated the secreted abnormalities seen in serum N-glycosylation, and a portion of the mutant protein appears to relocate to a distinct, non-Golgi compartment, possibly ER exit sites. These cells also show a gene dosage-dependent alteration in the Golgi morphology and reduced intraluminal pH that may account for the altered glycosylation. In summary, we identify a recurrent mutation in SLC37A4 that causes a dominantly inherited congenital disorder of glycosylation characterized by coagulopathy and liver dysfunction with abnormal serum N-glycans.
Keywords: Golgi pH; coagulopathy; congenital disordes of glycosylation; exome sequencing; glycosylation.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
SLC37A4-CDG: New biochemical insights for an emerging congenital disorder of glycosylation with major coagulopathy.Clin Chim Acta. 2021 Oct;521:104-106. doi: 10.1016/j.cca.2021.07.005. Epub 2021 Jul 8. Clin Chim Acta. 2021. PMID: 34245688 Free PMC article.
-
CRISPR/Cas9 genome editing of SLC37A4 gene elucidates the role of molecular markers of endoplasmic reticulum stress and apoptosis in renal involvement in glycogen storage disease type Ib.Gene. 2019 Jun 30;703:17-25. doi: 10.1016/j.gene.2019.04.002. Epub 2019 Apr 3. Gene. 2019. PMID: 30951856
-
[Mutation in the SLC37A4 gene of glycogen storage disease type Ib in 15 families of the mainland of China].Zhonghua Er Ke Za Zhi. 2011 Mar;49(3):203-8. Zhonghua Er Ke Za Zhi. 2011. PMID: 21575371 Chinese.
-
Three novel SLC37A4 variants in glycogen storage disease type 1b and a literature review.J Int Med Res. 2023 Dec;51(12):3000605231216633. doi: 10.1177/03000605231216633. J Int Med Res. 2023. PMID: 38087503 Free PMC article. Review.
-
The SLC37 family of phosphate-linked sugar phosphate antiporters.Mol Aspects Med. 2013 Apr-Jun;34(2-3):601-11. doi: 10.1016/j.mam.2012.05.010. Mol Aspects Med. 2013. PMID: 23506893 Free PMC article. Review.
Cited by
-
Hemostatic defects in congenital disorders of glycosylation.Res Pract Thromb Haemost. 2023 Mar 30;7(3):100142. doi: 10.1016/j.rpth.2023.100142. eCollection 2023 Mar. Res Pract Thromb Haemost. 2023. PMID: 37193126 Free PMC article.
-
Active site variants in STT3A cause a dominant type I congenital disorder of glycosylation with neuromusculoskeletal findings.Am J Hum Genet. 2021 Nov 4;108(11):2130-2144. doi: 10.1016/j.ajhg.2021.09.012. Epub 2021 Oct 14. Am J Hum Genet. 2021. PMID: 34653363 Free PMC article.
-
SLC37A4-CDG: New biochemical insights for an emerging congenital disorder of glycosylation with major coagulopathy.Clin Chim Acta. 2021 Oct;521:104-106. doi: 10.1016/j.cca.2021.07.005. Epub 2021 Jul 8. Clin Chim Acta. 2021. PMID: 34245688 Free PMC article.
-
Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: An update for 2021-2022.Mass Spectrom Rev. 2025 May-Jun;44(3):213-453. doi: 10.1002/mas.21873. Epub 2024 Jun 24. Mass Spectrom Rev. 2025. PMID: 38925550 Free PMC article. Review.
-
Metabolic Cardiomyopathies and Cardiac Defects in Inherited Disorders of Carbohydrate Metabolism: A Systematic Review.Int J Mol Sci. 2023 May 11;24(10):8632. doi: 10.3390/ijms24108632. Int J Mol Sci. 2023. PMID: 37239976 Free PMC article.
References
-
- Francisco R., Marques-da-Silva D., Brasil S., Pascoal C., Dos Reis Ferreira V., Morava E., Jaeken J. The challenge of CDG diagnosis. Mol. Genet. Metab. 2019;126:1–5. - PubMed
-
- Hamdan F.F., Myers C.T., Cossette P., Lemay P., Spiegelman D., Laporte A.D., Nassif C., Diallo O., Monlong J., Cadieux-Dion M., Deciphering Developmental Disorders Study High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am. J. Hum. Genet. 2017;101:664–685. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
