

Leveraging both individual-level genetic data and GWAS summary statistics increases polygenic prediction
- PMID: 33964208
- PMCID: PMC8206385
- DOI: 10.1016/j.ajhg.2021.04.014
Leveraging both individual-level genetic data and GWAS summary statistics increases polygenic prediction
Abstract
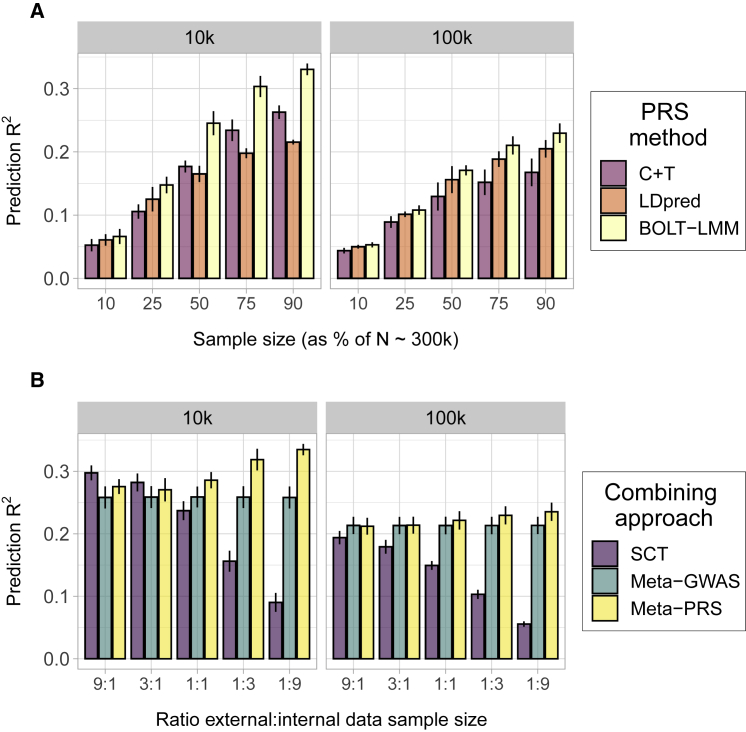
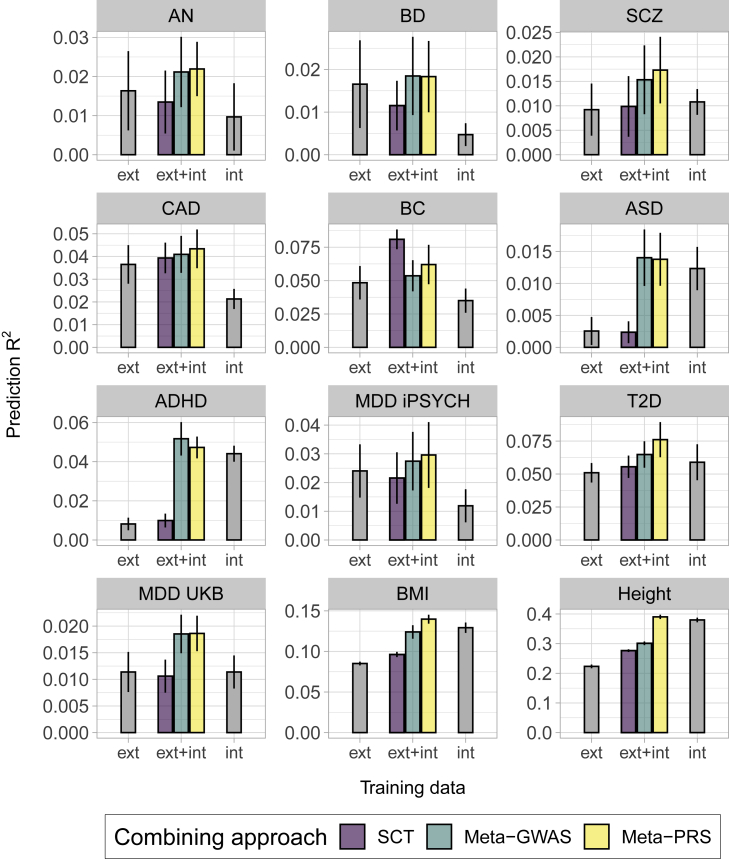
The accuracy of polygenic risk scores (PRSs) to predict complex diseases increases with the training sample size. PRSs are generally derived based on summary statistics from large meta-analyses of multiple genome-wide association studies (GWASs). However, it is now common for researchers to have access to large individual-level data as well, such as the UK Biobank data. To the best of our knowledge, it has not yet been explored how best to combine both types of data (summary statistics and individual-level data) to optimize polygenic prediction. The most widely used approach to combine data is the meta-analysis of GWAS summary statistics (meta-GWAS), but we show that it does not always provide the most accurate PRS. Through simulations and using 12 real case-control and quantitative traits from both iPSYCH and UK Biobank along with external GWAS summary statistics, we compare meta-GWAS with two alternative data-combining approaches, stacked clumping and thresholding (SCT) and meta-PRS. We find that, when large individual-level data are available, the linear combination of PRSs (meta-PRS) is both a simple alternative to meta-GWAS and often more accurate.
Keywords: PRS; complex traits; genetic prediction; meta-analysis; polygenic risk scores; psychiatric disorders.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
C.M.B. reports: Shire (grant recipient, Scientific Advisory Board member); Idorsia (consultant); Lundbeckfonden (grant recipient); Pearson (author, royalty recipient). The other authors declare no competing interests.
Figures
References
-
- Wray N.R., Lee S.H., Mehta D., Vinkhuyzen A.A., Dudbridge F., Middeldorp C.M. Research review: Polygenic methods and their application to psychiatric traits. J. Child Psychol. Psychiatry. 2014;55:1068–1087. - PubMed
-
- Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D1012. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
