

The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity
- PMID: 33965003
- PMCID: PMC8102044
- DOI: 10.1016/S2213-2600(21)00218-6
The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity
Abstract
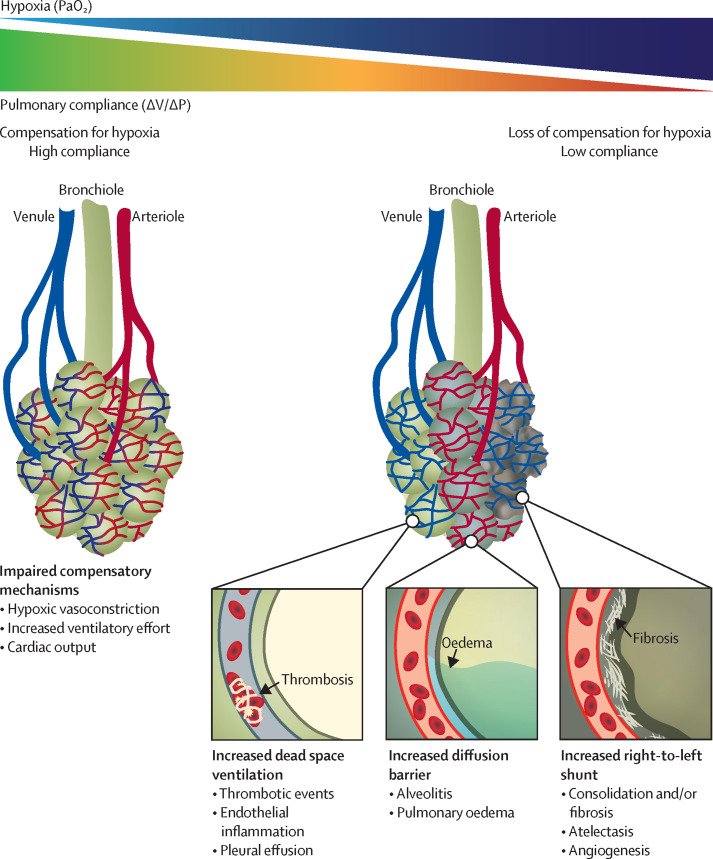
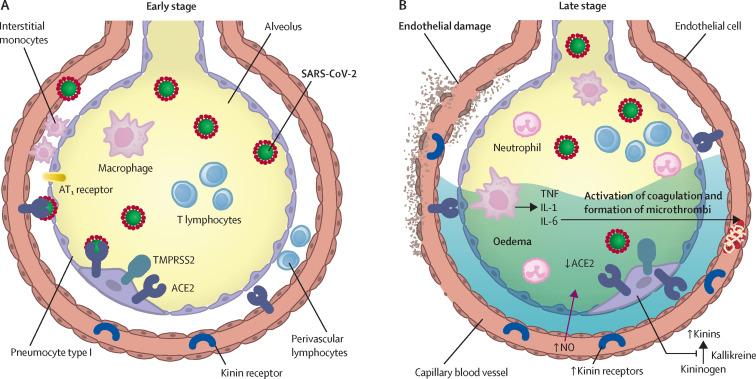
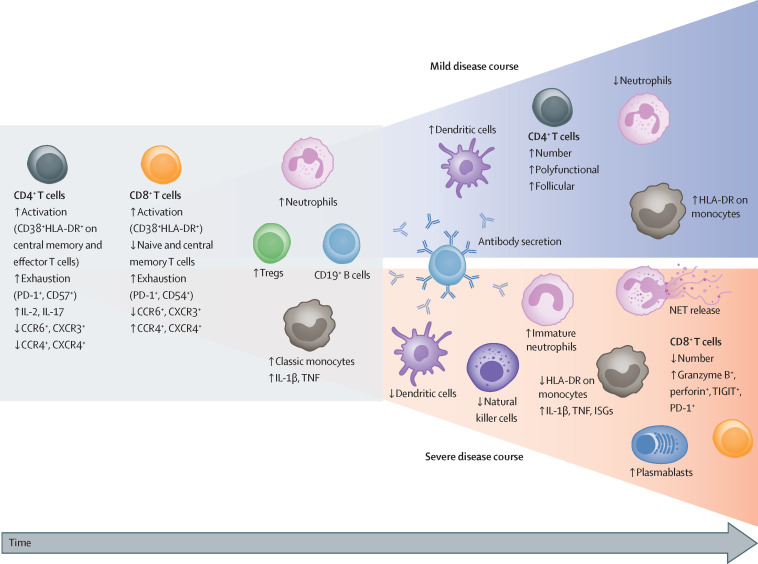
The zoonotic SARS-CoV-2 virus that causes COVID-19 continues to spread worldwide, with devastating consequences. While the medical community has gained insight into the epidemiology of COVID-19, important questions remain about the clinical complexities and underlying mechanisms of disease phenotypes. Severe COVID-19 most commonly involves respiratory manifestations, although other systems are also affected, and acute disease is often followed by protracted complications. Such complex manifestations suggest that SARS-CoV-2 dysregulates the host response, triggering wide-ranging immuno-inflammatory, thrombotic, and parenchymal derangements. We review the intricacies of COVID-19 pathophysiology, its various phenotypes, and the anti-SARS-CoV-2 host response at the humoral and cellular levels. Some similarities exist between COVID-19 and respiratory failure of other origins, but evidence for many distinctive mechanistic features indicates that COVID-19 constitutes a new disease entity, with emerging data suggesting involvement of an endotheliopathy-centred pathophysiology. Further research, combining basic and clinical studies, is needed to advance understanding of pathophysiological mechanisms and to characterise immuno-inflammatory derangements across the range of phenotypes to enable optimum care for patients with COVID-19.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests MSW has received unrestricted funding from Sartorius. GL reports personal fees from Swedish Orphan Biovitrum. TSp and JCS disclose institutional funding (received by the University of Bern) from Orion Pharma, Abbott Nutrition International, B Braun Medical, CSEM, Edwards Lifesciences, Kenta Biotech, Maquet Critical Care, Omnicare Clinical Research, Nestlé, Pierre Fabre Pharma, Pfizer, Bard Medica, Abbott, Anandic Medical Systems, PanGas Healthcare, Bracco, Hamilton Medical, Fresenius Kabi, Getinge Group Maquet, Dräger, Teleflex Medical, GlaxoSmithKline, Merck Sharp and Dohme, Eli Lilly, Baxter, Astellas, AstraZeneca, CSL Behring, Novartis, Covidien, Nycomed, Phagenesis, and Hemotune. MGN reports grants from GlaxoSmithKline and ViiV Healthcare, and was a scientific founder of TTxD. All other authors declare no competing interests.
Figures
Comment in
-
COVID-19 pathophysiology: looking beyond acute disease.Lancet Respir Med. 2021 Jun;9(6):545. doi: 10.1016/S2213-2600(21)00242-3. Lancet Respir Med. 2021. PMID: 34089668 Free PMC article. No abstract available.
References
-
- Worldometer COVID-19 coronavirus pandemic. April 25, 2021. https://www.worldometers.info/coronavirus/
-
- Johns Hopkins University of Medicine COVID-19 dashboard by the Center for Systems Science and Engineering (CSS) at Johns Hopkins University (JHU) https://coronavirus.jhu.edu/map.html
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
