

Refinement of α-Synuclein Ensembles Against SAXS Data: Comparison of Force Fields and Methods
- PMID: 33968988
- PMCID: PMC8100456
- DOI: 10.3389/fmolb.2021.654333
Refinement of α-Synuclein Ensembles Against SAXS Data: Comparison of Force Fields and Methods
Abstract
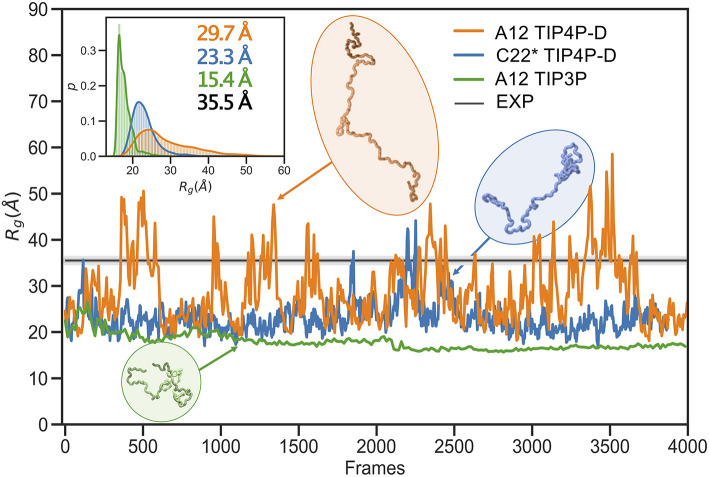
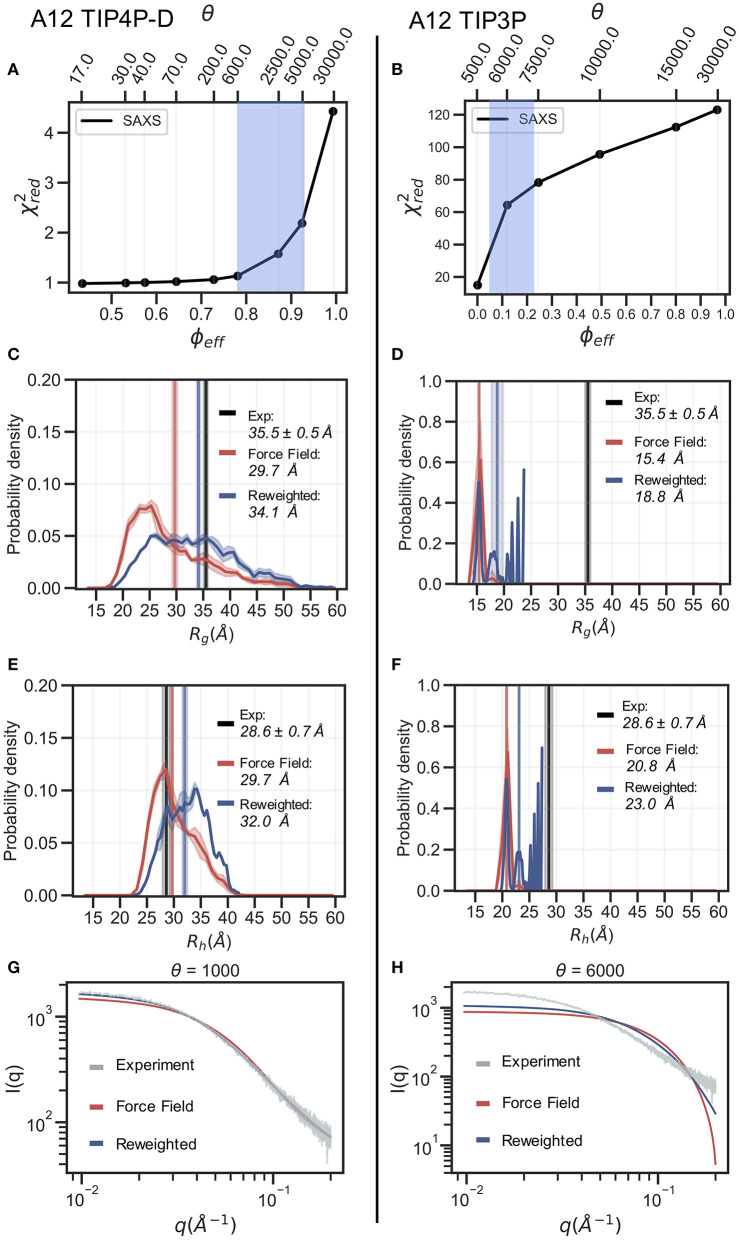
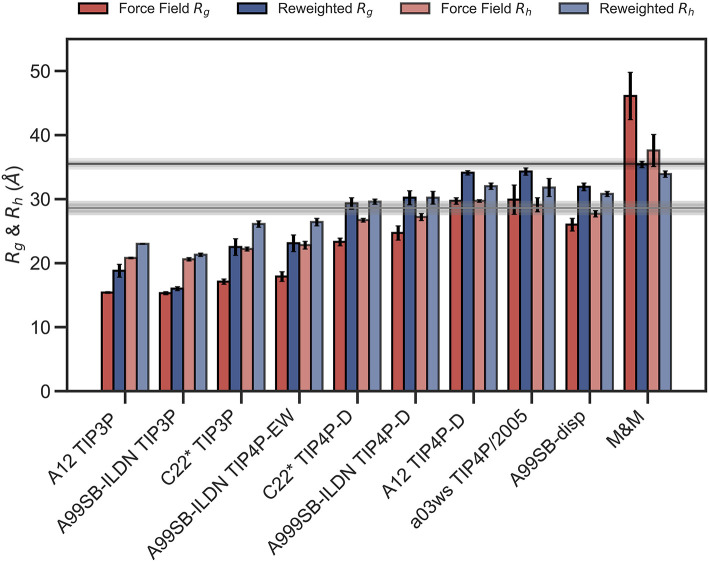
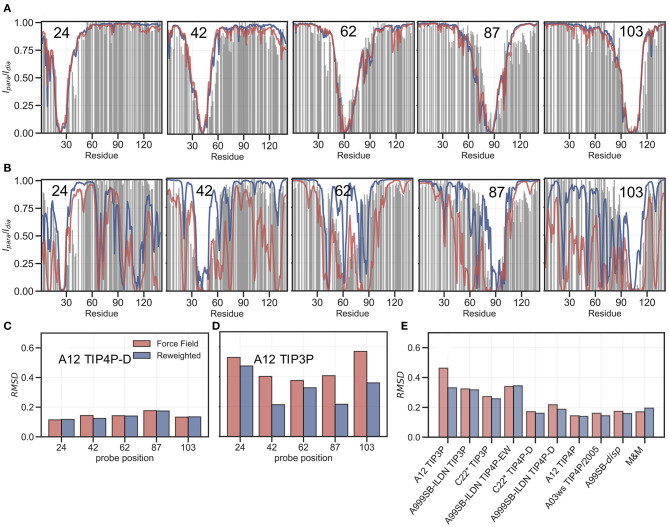
The inherent flexibility of intrinsically disordered proteins (IDPs) makes it difficult to interpret experimental data using structural models. On the other hand, molecular dynamics simulations of IDPs often suffer from force-field inaccuracies, and long simulation times or enhanced sampling methods are needed to obtain converged ensembles. Here, we apply metainference and Bayesian/Maximum Entropy reweighting approaches to integrate prior knowledge of the system with experimental data, while also dealing with various sources of errors and the inherent conformational heterogeneity of IDPs. We have measured new SAXS data on the protein α-synuclein, and integrate this with simulations performed using different force fields. We find that if the force field gives rise to ensembles that are much more compact than what is implied by the SAXS data it is difficult to recover a reasonable ensemble. On the other hand, we show that when the simulated ensemble is reasonable, we can obtain an ensemble that is consistent with the SAXS data, but also with NMR diffusion and paramagnetic relaxation enhancement data.
Keywords: NMR; intrinsically disordered protein; molecular dynamics simulation; protein; small-angle X-ray scattering.
Copyright © 2021 Ahmed, Skaanning, Jussupow, Newcombe, Kragelund, Camilloni, Langkilde and Lindorff-Larsen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., et al. . (2015). Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. 10.1016/j.softx.2015.06.001 - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources