

DNMT3A haploinsufficiency causes dichotomous DNA methylation defects at enhancers in mature human immune cells
- PMID: 33970190
- PMCID: PMC8111463
- DOI: 10.1084/jem.20202733
DNMT3A haploinsufficiency causes dichotomous DNA methylation defects at enhancers in mature human immune cells
Abstract
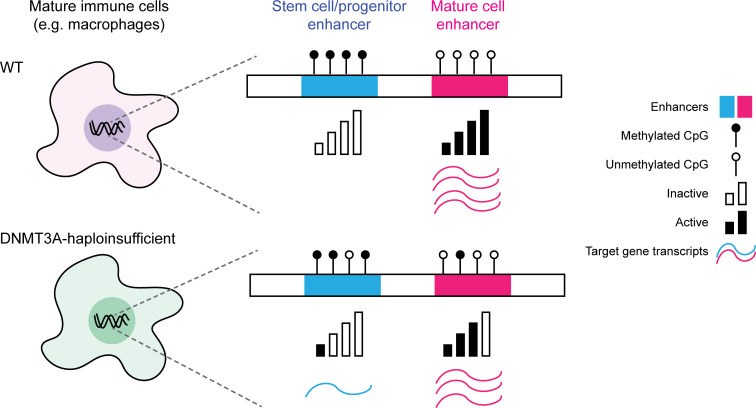
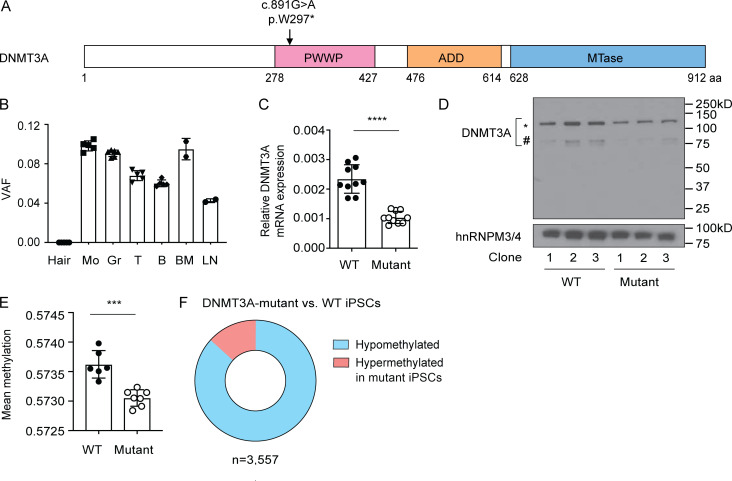
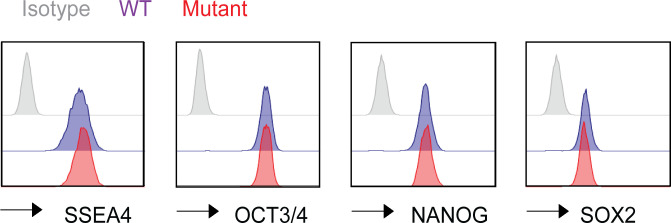
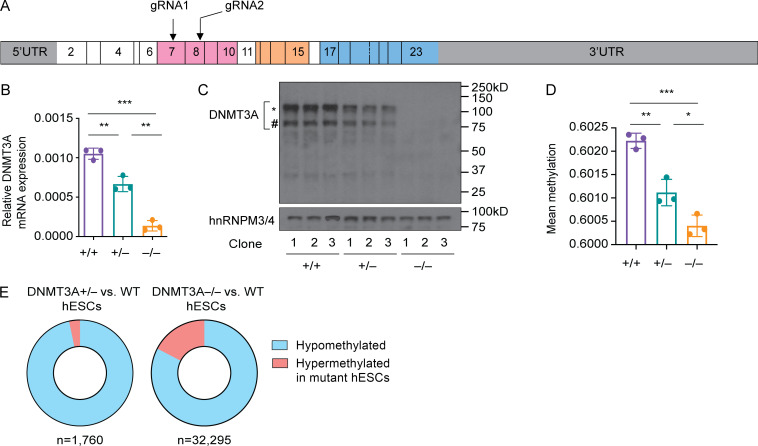
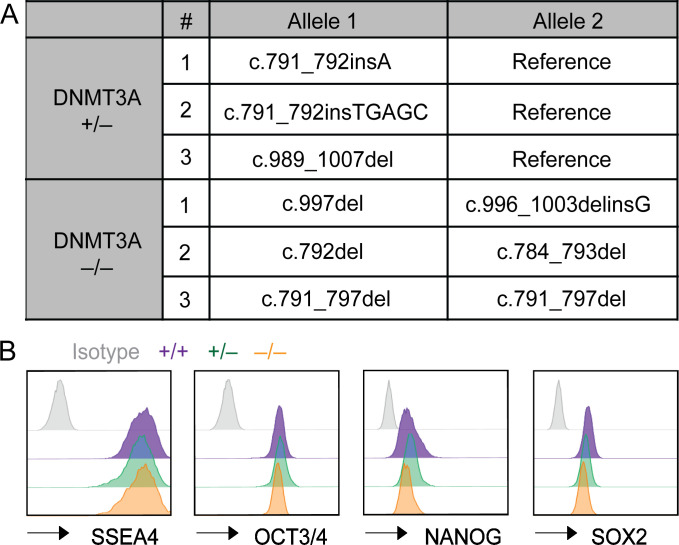
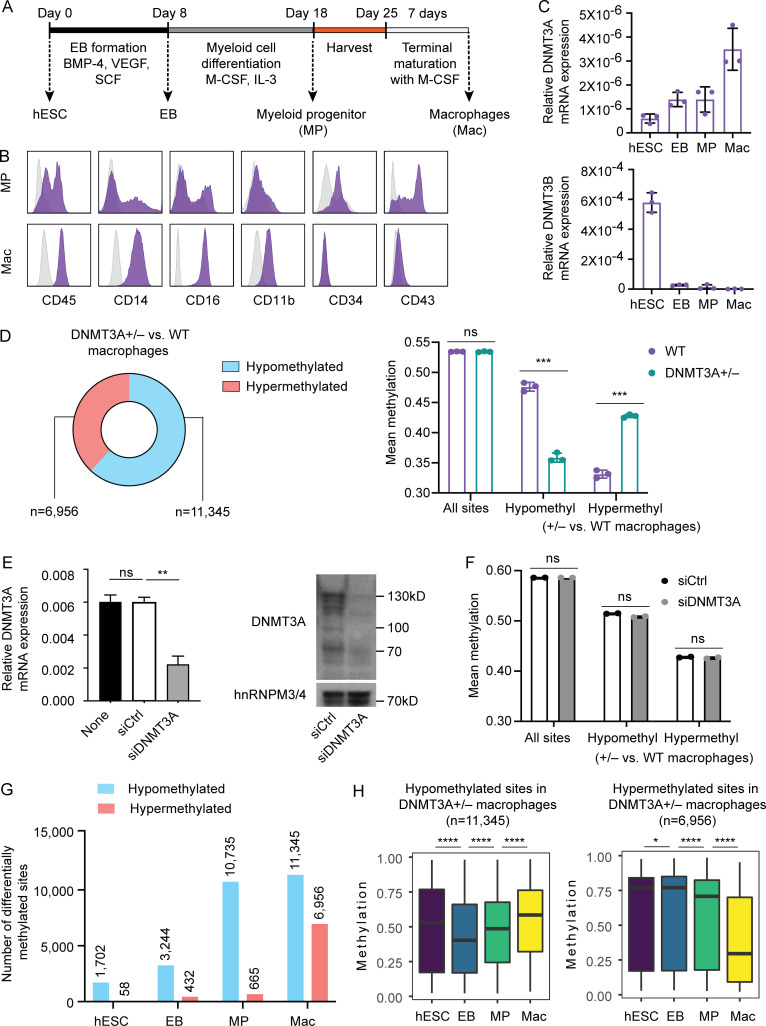
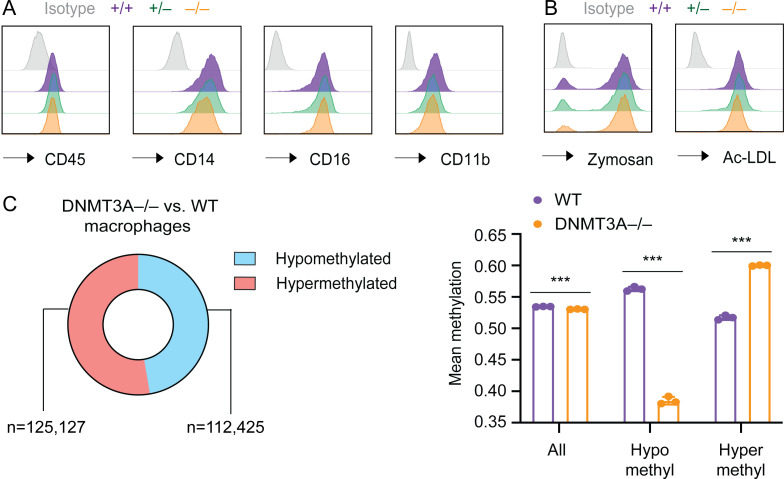
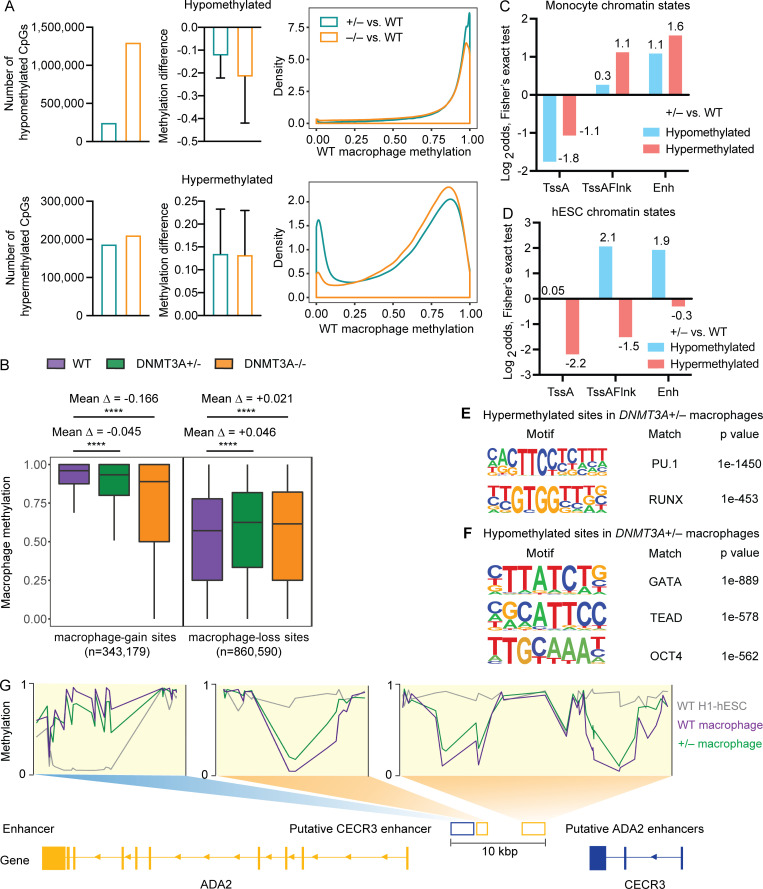
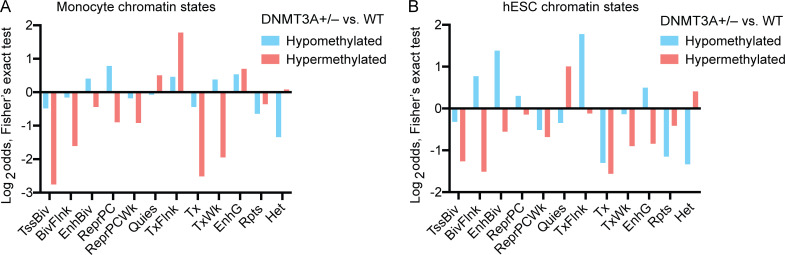
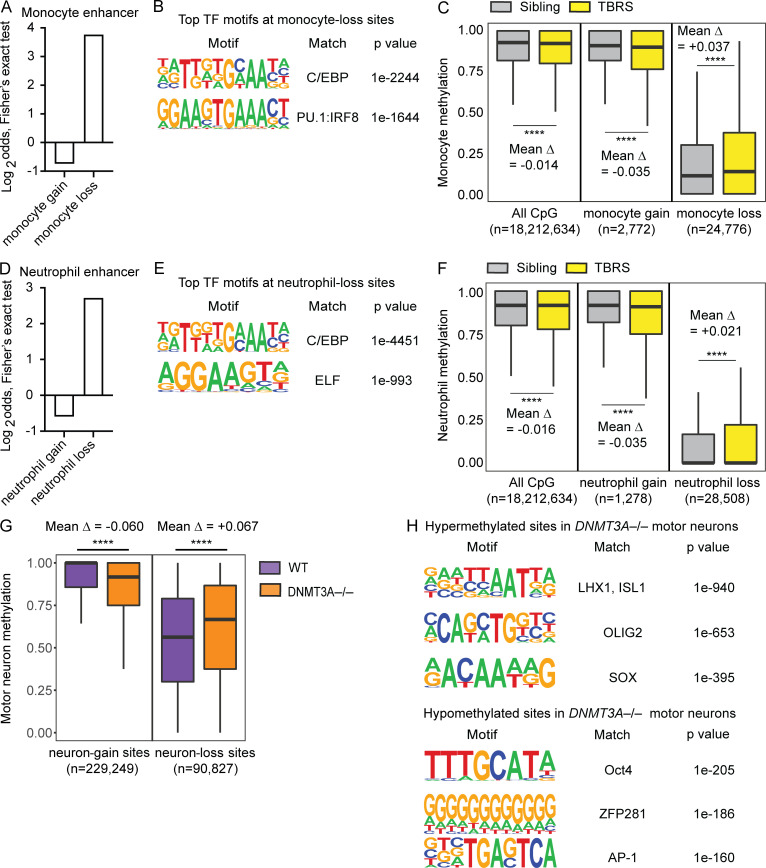
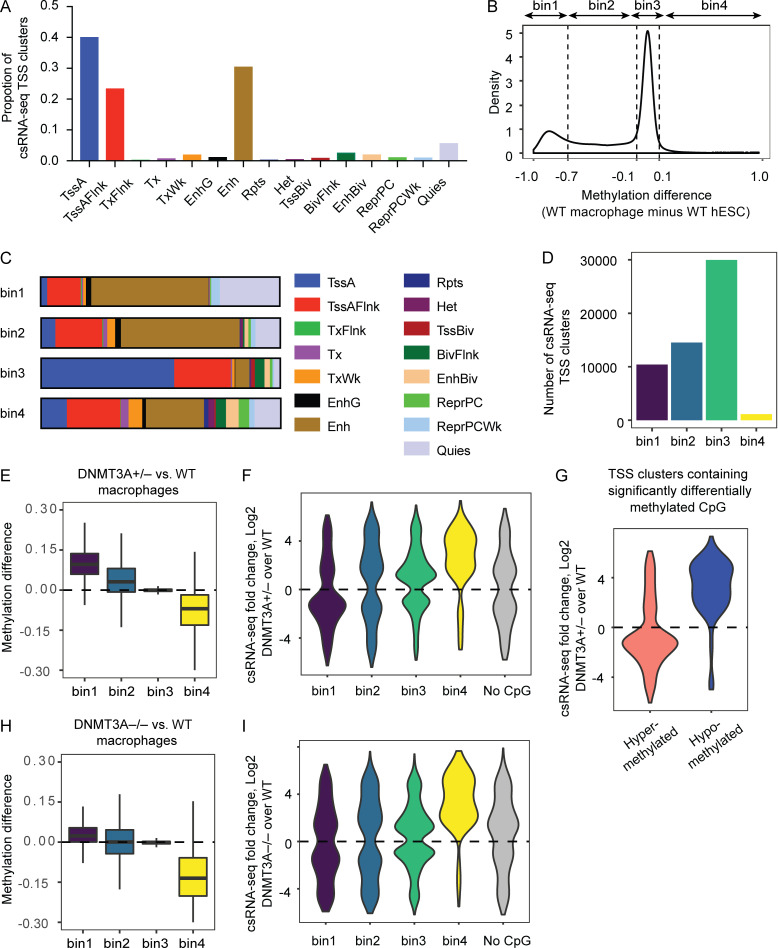
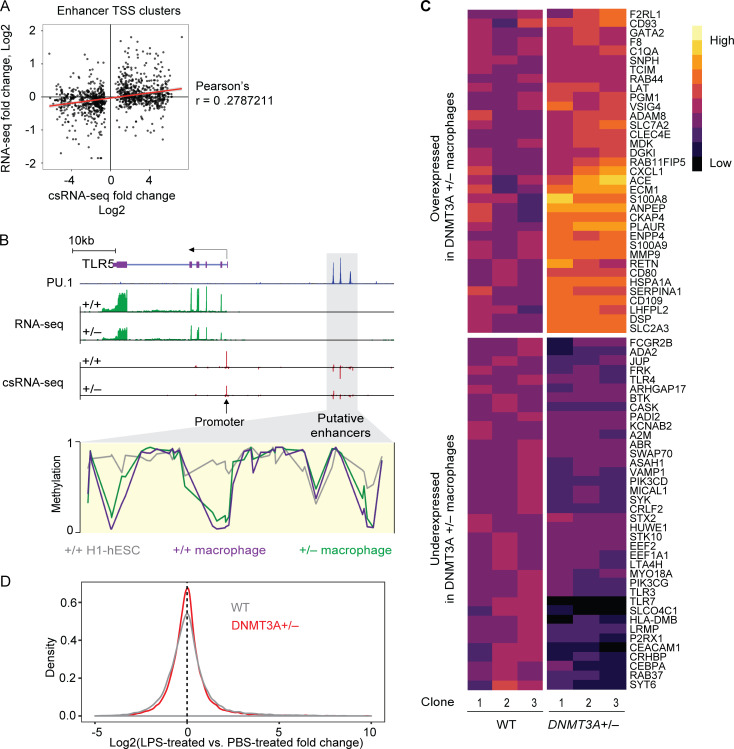
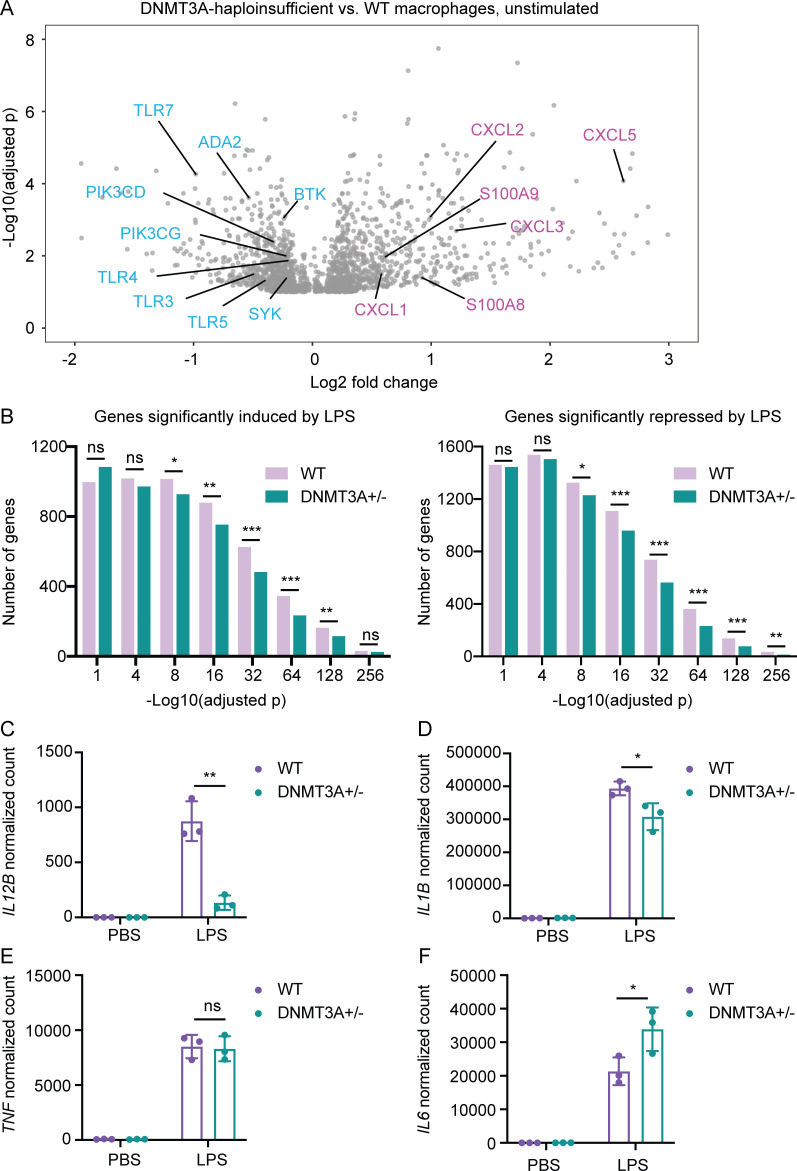
DNMT3A encodes an enzyme that carries out de novo DNA methylation, which is essential for the acquisition of cellular identity and specialized functions during cellular differentiation. DNMT3A is the most frequently mutated gene in age-related clonal hematopoiesis. As such, mature immune cells harboring DNMT3A mutations can be readily detected in elderly persons. Most DNMT3A mutations associated with clonal hematopoiesis are heterozygous and predicted to cause loss of function, indicating that haploinsufficiency is the predominant pathogenic mechanism. Yet, the impact of DNMT3A haploinsufficiency on the function of mature immune cells is poorly understood. Here, we demonstrate that DNMT3A haploinsufficiency impairs the gain of DNA methylation at decommissioned enhancers, while simultaneously and unexpectedly impairing DNA demethylation of newly activated enhancers in mature human myeloid cells. The DNA methylation defects alter the activity of affected enhancers, leading to abnormal gene expression and impaired immune response. These findings provide insights into the mechanism of immune dysfunction associated with clonal hematopoiesis and acquired DNMT3A mutations.
© 2021 Lim et al.
Conflict of interest statement
Disclosures: D.C. Fajgenbaum reported grants from EUSA Pharma and non-financial support from Pfizer outside the submitted work; in addition, D.C. Fajgenbaum had a patent for "Methods of Treating Idiopathic Multicentric Castleman Disease with JAK1/2 inhibition" pending (no licensee) and a patent for "Treatment of Castleman Disease" pending (no licensee). No other disclosures were reported.
Figures
References
-
- Aryee, M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., and Irizarry R.A.. 2014. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 30:1363–1369. 10.1093/bioinformatics/btu049 - DOI - PMC - PubMed
-
- Baker, T.S., Gambino K.J., Schriefer L., Lim J.Y., Steinberg K.M., Fajgenbaum D.C., Martín García-Sancho A., and Byun M.. 2018. A novel FAS mutation with variable expressivity in a family with unicentric and idiopathic multicentric Castleman disease. Blood Adv. 2:2959–2963. 10.1182/bloodadvances.2018023911 - DOI - PMC - PubMed
-
- Bock, C., Kiskinis E., Verstappen G., Gu H., Boulting G., Smith Z.D., Ziller M., Croft G.F., Amoroso M.W., Oakley D.H., et al. 2011. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 144:439–452. 10.1016/j.cell.2010.12.032 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
