

Infection-induced inflammation from specific inborn errors of immunity to COVID-19
- PMID: 33971084
- PMCID: PMC8236961
- DOI: 10.1111/febs.15961
Infection-induced inflammation from specific inborn errors of immunity to COVID-19
Abstract
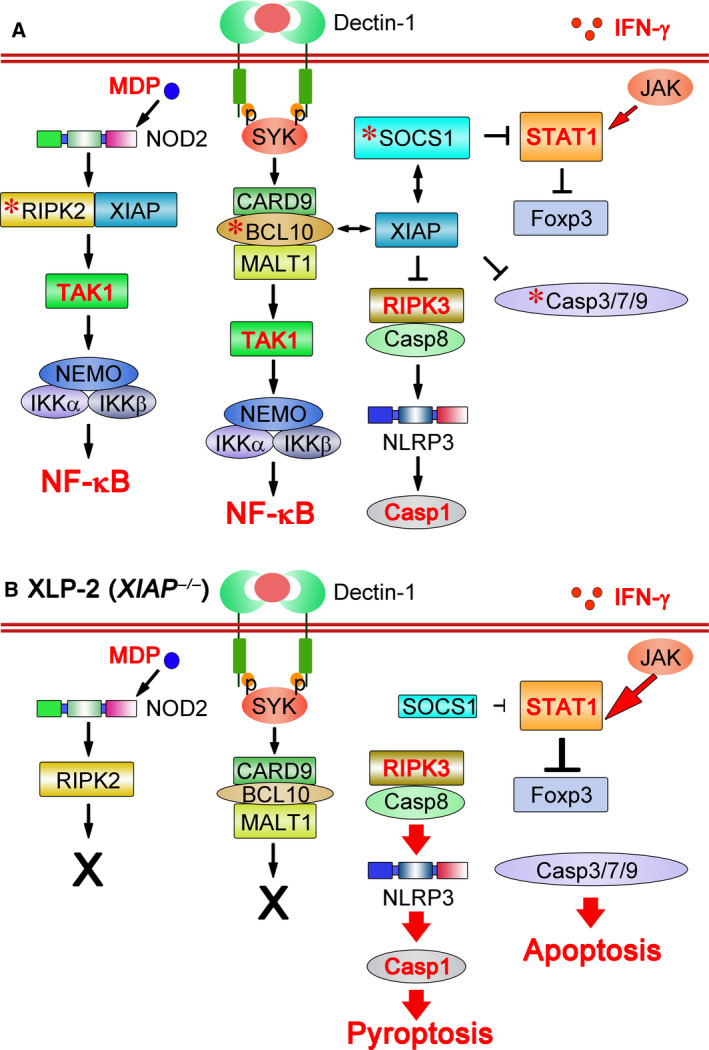
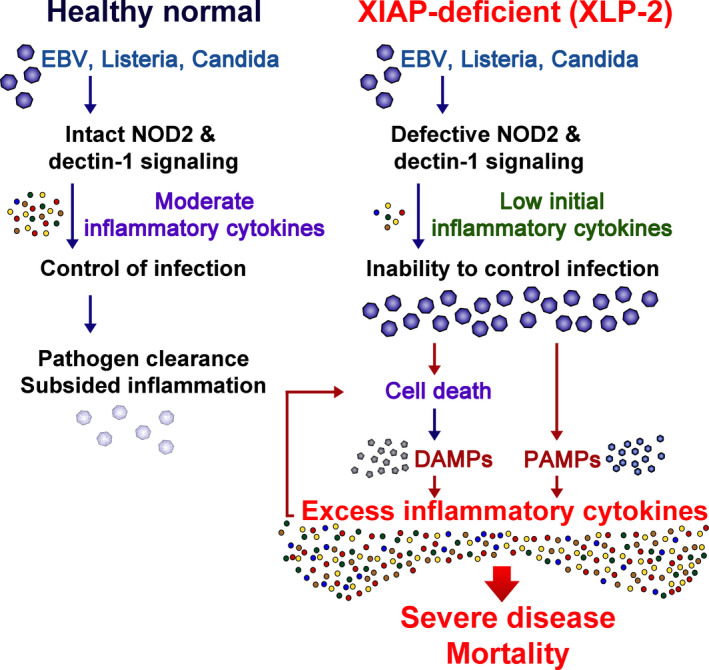
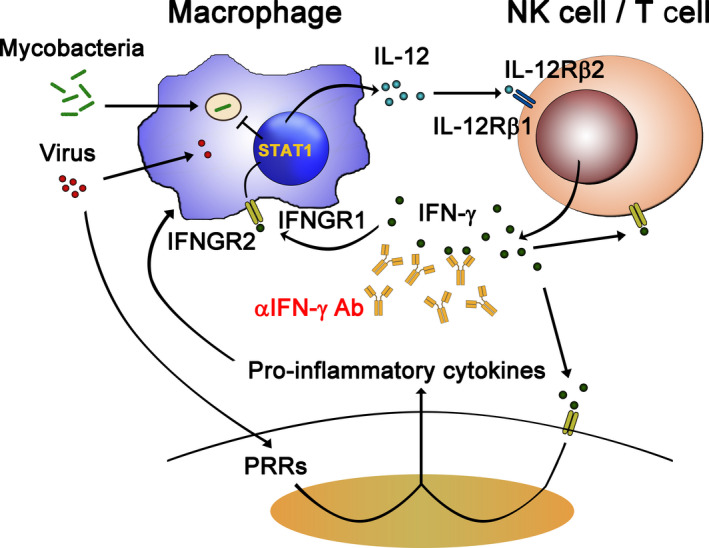
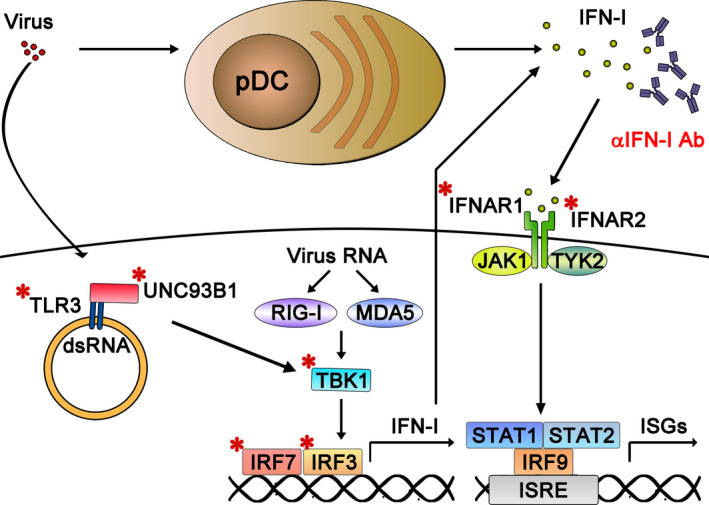
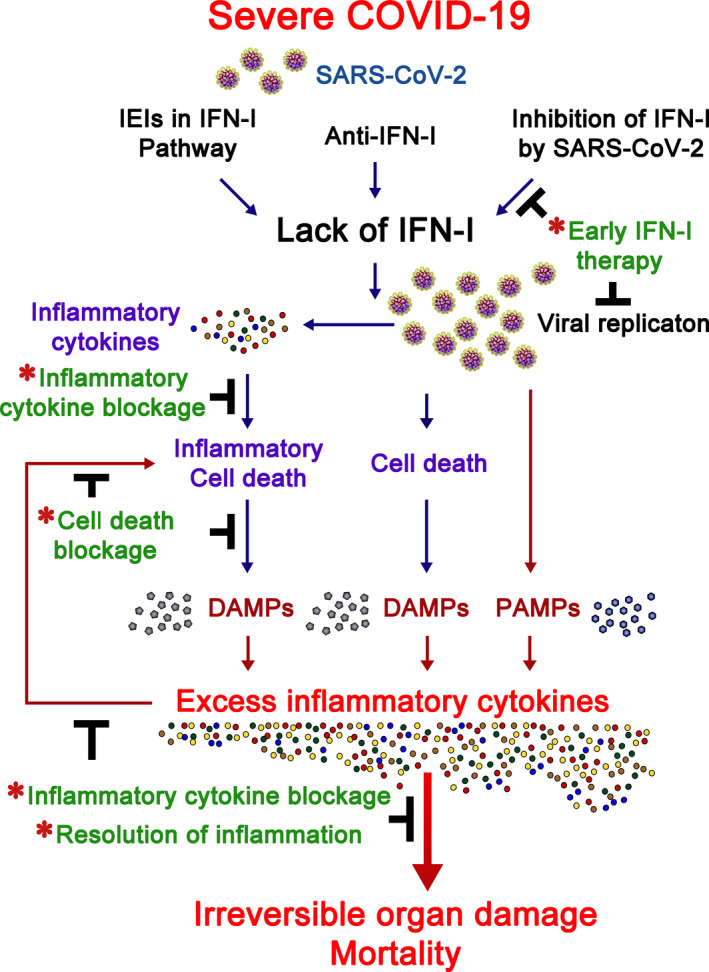
Inborn errors of immunity (IEIs) are a group of genetically defined disorders leading to defective immunity. Some IEIs have been linked to mutations of immune receptors or signaling molecules, resulting in defective signaling of respective cascades essential for combating specific pathogens. However, it remains incompletely understood why in selected IEIs, such as X-linked lymphoproliferative syndrome type 2 (XLP-2), hypo-immune response to specific pathogens results in persistent inflammation. Moreover, mechanisms underlying the generation of anticytokine autoantibodies are mostly unknown. Recently, IEIs have been associated with coronavirus disease 2019 (COVID-19), with a small proportion of patients that contract severe COVID-19 displaying loss-of-function mutations in genes associated with type I interferons (IFNs). Moreover, approximately 10% of patients with severe COVID-19 possess anti-type I IFN-neutralizing autoantibodies. Apart from IEIs that impair immune responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), SARS-CoV-2 encodes several proteins that suppress early type I IFN production. One primary consequence of the lack of type I IFNs during early SARS-CoV-2 infection is the increased inflammation associated with COVID-19. In XLP-2, resolution of inflammation rescued experimental subjects from infection-induced mortality. Recent studies also indicate that targeting inflammation could alleviate COVID-19. In this review, we discuss infection-induced inflammation in IEIs, using XLP-2 and COVID-19 as examples. We suggest that resolving inflammation may represent an effective therapeutic approach to these diseases.
Keywords: COVID-19; SARS-CoV-2; XLP-2; anticytokine autoantibodies; inborn errors of immunity.
© 2021 Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Finkel Y, Mizrahi O, Nachshon A, Weingarten‐Gabbay S, Morgenstern D, Yahalom‐Ronen Y, Tamir H, Achdout H, Stein D, Israeli O et al. (2020) The coding capacity of SARS‐CoV‐2. Nature 589, 125–130. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
